Research Article
Growth Kinetics of Rabies Virus in BHK-21 Cells Using Fluorescent Activated Cell Sorter (FACS) Analysis and a Monoclonal Antibody Based Cell- ELISA
Anandan Paldurai1#, Rabindra P. Singh1*, Praveen K Gupta2, Bhaskar Sharma3 and Kedar D. Pandey4
1Division of Biological Products, Indian Veterinary Research Institute, Izatnagar-243 122, Uttar Pradesh, India
2Division of Veterinary Biotechnology, Indian Veterinary Research Institute, Izatnagar-243 122, Uttar Pradesh, India
3Division of Biochemistry, Indian Veterinary Research Institute, Izatnagar-243 122, Uttar Pradesh, India
4A-129, Gail Apartments, Sector-62, Noida-201301, Uttar Pradesh, India
#Maryland Regional College of Veterinary Medicine, University of Maryland, College Park, MD 20742, USA
Corresponding author: Rabindra Prasad Singh, Division of Biological Products, Indian Veterinary Research Institute, Izatnagar-243 122, Uttar Pradesh, India; E-mail: rpsingh@dr.com, rpsingh@ivri.res.in
Citation: Paldurai A, Singh RP, Gupta PK, Sharma B, Pandey KD. Growth Kinetics of Rabies Virus in BHK-21 Cells Using Fluorescent Activated Cell Sorter (FACS) Analysis and a Monoclonal Antibody Based Cell-ELISA. J Immunol Vaccine Technol. 2014;1(1): 103.
Copyright © 2014 Rabindra P. Singh et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Immunology and Vaccine Technology | Volume: 1, Issue: 1
Submission: 08/08/2014; Accepted: 24/10/2014; Published: 27/10/2014
Reviewed & Approved by Dr. Sanjeev Shukla, AssistantProfessor, Department Of Biological Sciences, IndianInstitute of Science Education and Research Bhopal, India
Abstract
Introduction: Rabies is a highly fatal, neurotropic viral zoonosis of warm-blooded animals including man caused by rabies virusbelonging to genus Lyssavirus of the family Rhabdoviridae under order Mononegavirales. Now-a-days rabies cell culture vaccines arebeing used as a curative and prophylactic immunization in animals. The present investigation aimed at refinement/improvement of theexisting cell culture rabies vaccine protocol, envisaging critical investigation on kinetics of growth of Pasteur virus strain in BHK-21 clone13 cells.
Materials and Methods: Mouse monoclonal antibodies were generated against rabies virus for the development of a cell-ELISA as anin-process control assay. Pasteur virus strain of rabies virus was propagated in BHK-21 cells. Virus samples were collected at differenttime intervals for investigation of growth kinetics using cell-ELISA, FAT and fluorescence activated cell sorter analysis.
Results: The monoclonal antibody based cell-ELISA developed during the present study may serve as a useful assay with provenrepeatability for in-process monitoring of growth of rabies virus in BHK-21 cells. This assay although less sensitive, correlated well withFAT for the determination of optimal period of harvesting the virus. Fluorescence Activated Cell Sorter (FACS) analysis indicated itssuitability and higher level of sensitivity for monitoring of rabies virus propagation in BHK-21 cells. Growth pattern of the virus using allthe three tests indicated that virus has eclipse phase at around 9-12 h post infection. The study on rabies virus growth kinetics suggestedthat the optimum quantity of infectious virus particles could be harvested at around 48 h of infection using 0.1 multiplicity of infection ofrabies virus.
Conclusion: The findings of the present investigation if applied successfully are likely to impact the production protocol of cell culturerabies vaccine with the elimination of hurdles like concentration of virus and use of mice for vaccine virus titration, etc.
Keywords: Rabies virus; Growth Kinetics; Fluorescent Activated Cell Sorter (FACS) analysis; Cell-ELISA; FAT
Introduction
Rabies is a severe and fatal viral disease of the central nervous system of warm-blooded animals, including man [1]. It is enzootic and sometimes epizootic in a variety of mammalian species, insectivorousand hematophagous bats [2]. The causative rabies virus is usually introduced by a bite wound, although penetration can occur through intact mucous membranes and the digestive tract [3], but not through intact skin. Airborne natural infection is also possible in exceptionalcircumstances, for example in caves harbouring large numbers of bats carrying the virus [4]. It has remained an important cause of disease in animal reservoirs with significant human cases, especially in the developing countries [5]. Rabies exposure has profound worldwidemedical and economic implications [6], with as many as 4 million people per year receiving post-exposure treatment to prevent rabies [7].
Rabies virus belongs to the genus: Lyssavirus, of the family: Rhabdoviridae under the order: Mononegavirales [8]. The virus replicates in the cytoplasm of a variety of cells and released by budding. Specific cellular receptors for rabies virus are characterized only in neurons [9]. Although the virus replicates in a variety of cell lines like baby hamster kidney cells (BHK-21), African green monkey cells (Vero), human diploid cells and other cell lines of non-neuronal origin, cytopathic changes are not observed in these cells [10]. Worktowards rabies diagnosis in the past concentrated on histopathological methods that exploited different staining processes for the detection of cytoplasmic virus inclusions, the Negri bodies [11]. Later, directimmunofluorescence [12] was used extensively and replaced staining techniques. In some cases, however virus isolation is necessary for which procedures used most frequently involve mice [13] or one of a number of different cell lines [14]. Other virus detection methods include enzyme-linked immunosorbent assay [15], monoclonal antibody (Ab) based virus neutralization test and florescent antibody test (FAT) [16], electron microscopy [17] and Polymerase Chain Reaction. Flowcytometry has also been described for monitoring rabies infection in BHK-21 and C6 cell lines [18].
Many different forms of rabies vaccine have been produced since Pasteur’s original success in 1882. A series of cell culture based vaccines against rabies is available from different manufacturers for use in the dogs. However, a similar vaccine to be used for thelarge animals (cattle and buffalo) is not very much authenticated in relation to its protective efficacy. Majority of the interpretations under such circumstances are made from the findings in pet animals. Standard, but cost effective production of rabies cell culture vaccine is a necessity. Since rabies virus is a non-cytopathogenic in cell culture, its quantitation requires extensive use of experimental animals (e.g. mice) at frequent intervals during the process of vaccine production [19]. Virus quantitation using a heterologous system does not givea clear picture of infectivity titre. Furthermore, the rabies virus is very thermolabile [7]. Therefore production and quality control of cell culture rabies vaccine requires simplified assays like ‘fixed cellmonolayer’ based enzyme-linked immunosorbent assay (cell-ELISA) for monitoring virus quantity in vaccine samples. The cell-ELISA has already been used for infectivity assay of some of the viruses likepeste des petits ruminants virus by us [20] and polio virus [21]. Due to low virus titre in the harvest, production of rabies vaccine involves frequent virus concentration to get sufficient antigenic value/mass[19]. Being a fatal zoonotic disease and the risks involved with the handling of virus, it is always desired to produce quality vaccine with a minimum downstream processing of the live virus. Furthermore, due to difficulties in virus quantitation, some batches of cell culture based rabies vaccines result in low antigenic values. This indirectly affects protective efficacy of the vaccine. The anti-rabies vaccination in large ruminants in India and other developing countries is carried out mainly as a therapeutic measure following a post bite vaccination schedule subsequent upon a dog bite that to without rabies immunoglobulin, which is a costly affair. Under these circumstances the antigenic value of such a vaccine and quality assurance is of utmost importance. This warrants no compromise at the level of vaccine quality. Therefore vaccine production protocol for such virus has to be very critical but simplified. Keeping all these points in mind the present study was planned with the objectives i) to develop an assay for in-process monitoring of rabies virus (Pasteur Virus strain) replication in BHK-21 clone 13 cells, ii) to determine the replication pattern of the virus in BHK-21 clone 13 cells with an idea for optimal antigen harvesting.
Materials and Methods
Cell lines, Virus, Antibodies, Antibody conjugates
The cell lines (BHK-21 clone 13 cells and Myeloma cells) used in the present study was received from the Division of Virology, Mukteswar campus, IVRI. BHK-21 clone 13 cells between 70 to 90 passages were used as the substrate for the growth of Pasteur virus (PV) strain of rabies virus. BHK-21 cells were propagated in GlasgowMinimum Essential Medium (GMEM) (G-6148, Sigma) containing10% fetal calf serum (FCS). Myeloma cells (SP2-O) were propagated and maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 20% FCS (Sigma, F-7524) under carbon dioxide tension in a desiccator under humid condition. These myeloma cells were used as fusion partners with sensitized spleen lymphocytes for hybridomaproduction.
PV strain of rabies virus having a titre of 4.5×105 focus forming units (FFU)/ml was propagated in BHK-21 cells. A harvest with an infective virus titre of 3×106 tissue culture infective dose 50 (TCID)50/ml was obtained and used for further investigation. Rabies virus was stored in aliquots at -70 ºC.
Rabbit anti-rabies (whole virus) serum was kindly provided by Dr. P.K. Gupta, Senior Scientist, Division of Veterinary Biotechnology, IVRI for the use in the development of a polyclonal antibody basedcell-ELISA. mAb from the rabies virus specific hybridoma clone ‘Rab- 48’ was raised in the present study and used as the primary antibody in the development of a mAb based cell-ELISA and investigation of growth kinetics.
Liquid Rabies anti-nucleocapsid fluorescein isothiocyanate (FITC) conjugate (357-2114, Bio-Rad) was used for FAT and for the fluorescence activated cell sorter (FACS) analysis to detect and/or quantify virus infected cells for the determination of rabies virusgrowth kinetics in BHK-21 cells. Goat anti-Rabbit horseradish peroxidase (HRP) conjugate (HPO3, Bangalore Genei) was used a secondary antibody in the polyclonal antibody based cell-ELISA. Goat anti-mouse horseradish peroxidase (HRP) conjugate was used as the secondary antibody in mAb based cell-ELISA.
Propagation of virus
Rabies virus (PV strain) was propagated in BHK-21 clone 13 cells. Using 0.1 MOI, rabies virus was co-cultivated with BHK-21 cells at 37 ºC for 72 h in a 75 cm2 cell culture flask. During the course of virus production, pH was adjusted to around 7.6 using 7.5% sodium bicarbonate solution. After 72 h, virus was harvested and frozen at -70 ºC. Virus harvest was thawed and centrifuged at 2000 rpm for 10 min. Aliquot of the cell free virus was stored at -70 ºC. Virus witha titre of 3×106 TCID50/ml was used in the present study for further investigation.
Production of mAbs
Two Swiss-albino mice (weighing 16-20 g, 3-4 weeks age of either sex) were immunized using rabies virus antigen. This included primary immunization of mice using crude antigen followed by two booster doses of vaccine on day 42 and 55 and fusion on day 56. Myeloma cells (SP2-O) and feeder cells (peritoneal macrophages) were prepared as per the protocol already described [22,23]. Well towell screening of hybridoma clones was performed at appropriate stage of the growth of the clones. A cell-ELISA assay system [21] was employed for screening of rabies specific mAbs in the hybridoma supernatants using acetone fixed rabies virus infected and mock infected BHK-21 cells. The hybridoma culture supernatants whichgave at least two times the A492 values with infected BHK-21cells as compared to mock infected cells were considered positive for rabies virus. Positive clones were amplified from 96-well cell culture plates to 24-well cell culture plates and subsequently to 25 cm2 cell culture flasks. Single cell cloning and sub-cloning of rabies virus positive hybridoma were performed using limiting dilution method [24,25]. Plates were observed for the growth of cells regularly. They were screened at appropriate growth stage (25% well surface) using cell-ELISA as described earlier. Single cell cloning was repeated twice so as to prove monoclonality of the hybridoma culture supernatants. Hybridoma clones derived from single cells were selected, expanded and preserved in -70 °C. Sufficient amount of mAbs were produced and used for cell-ELISA for determination of the growth pattern of rabies virus in BHK-21 cells.
Development and standardization of a mAb based Cell-ELISA
mAb derived from clone ‘Rab-48’ was employed to develop a cell-ELISA. The procedure followed for cell-ELISA is described in brief. Serially log10 diluted PV strain was co-cultivated with BHK-21 cells using 96-well cell culture plate keeping four replicates for each virusdilution. After 72 h of incubation at 37 ºC, spent medium was aspirated completely and cell monolayer was fixed with 100 μl chilled acetone(80:20 in PBS) per well and kept at -20 ºC for 1 h. Cell monolayer was air dried for 20-30 min in a laminar airflow. After air drying, the platewas blocked using 200 μl of blocking buffer (Gelatin-1%, FCS-0.5% and Tween 20-0.1% in PBS) and incubated at 37 ºC for 1 h or +4 ºC overnight. The blocking buffer was discarded and 100 μl of monoclonal antibody (1: 2 dilution of Rab-48) was added and plate was incubated at 37 ºC for 1 h. Plate was washed 3 times with wash buffer (0.01%Tween-20 in 0.0025M). 100 μl of rabbit-anti-mouse HRP conjugate was added per well (1:1000 dilution in blocking buffer) and incubated at 37 ºC for 1 h. Plate was washed 3 times with wash buffer. 100 μl of substrate-chromogen mixture (4 μl of 3% Hydrogen peroxide [H2O2] per ml of pre-warmed ortho-phenylene diamine [OPD]) was addedper well and the plate was observed for colour development. After the colour development, the reaction was stopped by adding 100 μl of 1M Sulphuric acid (H2SO4) per well and reading was taken at 492 nm in an ELISA reader. Based on the cell-ELISA reading, virus titrewas calculated using Reed and Muench formula [26] and expressed as TCID50/ml.
Titration of rabies virus using Cell-ELISA
Virus dilution was made using deep-well plate. A serial 10 fold dilutions of virus were made up to 10-7 dilutions. BHK-21 cells were taken at the final cell concentration of 4-6x105 cells per ml in 10%GMEM in a cell culture trough. From the deep-well plate, 100 μl of the log10 diluted virus samples were added to the cells in four replicates in the same order from 10-1 to 10-7, using a multi-channel pipette. 96-well cell culture plate was placed in a humidified desiccator and incubated at 37 ºC for 48 h. After 48 h, the spent medium was discarded and 2% GMEM was added at the rate of 100 μl per well. The cells were fixed after 72 h using chilled acetone and air dried for the application of cell-ELISA (as described above).
Investigation on virus growth kinetics
Rabies virus growth pattern was studied at 0.1 MOI in BHK-21 cells using FACS analysis, FAT and the newly developed mAb based cell-ELISA. The methodology adopted for the collection of virus samples for the investigation of rabies virus growth kinetics, virus titration, FAT and FACS is given below.
Preparation and collection of cell free and cell associated virus samples
Two 24 well cell culture plates were seeded with the BHK-21 cells uniformly at a cell concentration of 1.6×105 cells per well in GMEM-10% and incubated at 37 ºC in a humidified desiccator. At 75-80% confluency of the monolayer, cell count was determined from one well using haemocytometer method. Based on cell count 0.1 MOI ofPV strain was calculated and rabies virus diluted in 2% GMEM to get 0.1 MOI in 1 ml per well and likewise all wells were infected using 1 ml of virus dilution. Immediately after the infection of the BHK-21cell monolayers with 0.1 MOI of rabies virus, the supernatant was collected for zero ‘cell free’ sample in a sterile eppendorf tube. Then the cell monolayer was washed with 0.5 ml 2% GMEM and cells were collected in 1 ml of 2% GMEM by scrapping the monolayer. These samples were named as ‘cell associated’ virus sample for zero hr and stored at -70 ºC. Following the sample collection procedure as given above, the ‘cell free’ and ‘cell associated’ samples were collected at various time intervals viz. 6 h, 12 h, 18 h, 24 h, and after every 24h on 2nd day, 3rd day and up to 9th day. Each time after collection of samples, the 24 well plates were incubated at 37 ºC in a humidified desiccator, throughout this period the pH was adjusted to around 7.4 using 7.5% sodium bi-carbonate solution. The collected ‘cell free’ and‘cell associated’ rabies virus samples were titrated using cell-ELISA, FAT and FACS analysis for the evaluation of growth kinetics.
Titration of rabies virus samples for investigation ongrowth pattern
The ‘cell free’ and ‘cell associated’ virus samples collected for growth kinetics were thawed from -70 ºC and titrated using the mAb based cell-ELISA, FAT and FACS technique under identical conditions. The efficacy of the mAb based cell-ELISA was compared with the above said two tests for virus titration.
Cell-ELISA
The virus samples harvested for growth kinetics were thawed from -70º C and serial log10 dilutions were prepared for ‘cell free’ and ‘cell associated’ virus samples of each time interval starting from 0 hto 144 h in an ascending order. Virus samples were co-cultivated with BHK-21 cells using 96-well cell culture plates. With the first media change after 48 h, the plates were incubated up to 72 hrs, spent mediawas aspirated completely and cell monolayer was fixed with 100 μl chilled acetone (80:20 in PBS) per well and dried at -20 ºC for 1 h. The cell-ELISA was performed as described above.
Fluorescent Antibody Test (FAT)
The ‘cell free’ and ‘cell associated’ virus samples harvested at various time points were titrated with BHK-21 cells using cocultivation method. After the incubation period of 72 h, the medium was discarded completely and the cell monolayer was washed with phosphate buffer. Cells were fixed in chilled acetone (80:20 in PBS) at -20 ºC for 1 h. Acetone was aspirated completely and the platewas air dried rapidly. Rabies anti-nucleocapsid antibody conjugated with FITC (1:20) dilution in phosphate buffer, pH 7.2 was added at the rate of 25 μl per well to cover the monolayer entirely. The cell culture plate was incubated at 37 ºC for 30 min in a humid chamber.The plate was washed with phosphate buffer, pH 7.2 and rinsed in buffered glycerine (50% Glycerine in PBS, v/v). Plate was observed under fluorescent microscope and fluorescent foci were counted for the determination of virus titre. Virus titre was calculated using Reedand Muench formula [26] and expressed as FFU/ml.
Fluorescence Activated Cell Sorter (FACS) analysis
The virus samples harvested at various time intervals were titrated using FACS analysis following the protocol described earlier [18] with some modifications like, use of chilled acetone (80:20 in PBS) for fixing of cells and use of less quantity of anti-nucleocapsid FITC conjugate. The cell free and cell associated virus samples harvested at various time points were titrated in BHK-21 cells in 24-well cell culture plates by adsorption method on preformed 75-80% monolayer. After first media change at 48 hrs plates were incubated up to 72 h at 37 ºC in a desiccator. After 72 h, the supernatant was discarded completely and the cell monolayer was washed with phosphate buffer, pH 7.2. 300 μl of Trypsin-Versene (pre-warmed at 37 ºC) was added per well, when cells detached from surface, 700 μl of GMEM-10% was added and cells were pipetted to get single cells. Cells were collected in an Eppendorf tube and centrifuged at 2000 rpm for 10 minutes and the cell pellet was given a washing in 1 ml of phosphate buffer at 2000 rpm for 10 min. The cell pellets were dispersed gently to get single cells and cells were fixed in chilled acetone at -20 ºC for 1 h. After acetone fixation, cells were centrifuged at 2000 rpm for 10 min and acetone was discarded completely using separate tips for each dilution. The cell pellets were dispersed to single cells and were washed with PBS at 2000 rpm for 10 min and again the pellets were dispersed manually to get single cells. Liquid rabies anti-nucleocapsid FITC conjugate (1:20) dilution in phosphate buffer at pH 7.2 was added at the rateof 20 μl per eppendorf tubes only in 10-3 dilution all virus samples and incubated at 37 ºC for 30 min in a humid chamber. Cells were again washed with 1 ml Phosphate buffer (pH 7.2) and the cells were dispersed manually and re-suspended in 400 μl of Phosphate buffer and the samples were subjected for FACS analysis. Results of the FACS analysis was recorded as % of rabies virus infected cells(fluorescent cells) in each sample.
Cell-ELISA using various MOI of rabies virus
PV strain of rabies virus was co-cultivated at 0.01, 0.1 and 1.0 MOI with BHK-21 cells to determine optimal MOI and time of harvest for obtaining optimal virus titre. For virus titration, mAb based cell-ELISA was employed since it proved to be a convenient, easy to perform test with a considerable reliability. BHK-21 cells were seeded in three 25 cm2 cell culture flasks at the cell concentration of 2.4x106 cells per 5 ml in 10% GMEM. The cells were co-cultivated with 1 MOI, 0.1 MOI and 0.01 MOI of PV strain of rabies virus in the three flasks respectively the flasks were incubated at 37 ºC. At every 24 h interval after infection, the cell free virus was collected from each MOI flask separately and 5 ml of 2% GMEM pH 7.6 was added in each flask daily up to 144 h (6 days). ‘Cell free’ virus samples harvested from the 1 MOI, 0.1 MOI and 0.01 MOI flasks at 24 h interval were titrated on the same day in BHK-21 cells using co-cultivation method. mAb based cell-ELISA was performed (as described in section 2.6.2) in all the plates at the same time and readings were taken at 492 nm in an ELISA reader. Based on the ELISA reading, virus titre was calculated using Reed and Muench formula [26] and expressed as TCID50/ml..
Results
Generation of mouse hybridoma and development of a mAb cell-ELISA
Fusion of sensitized splenocytes and myeloma cells was performed for production of mouse hybridoma. The generation of mouse hybridoma clones against rabies virus antigen resulted in two useful clones for cell-ELISA, designated as ‘Rab-18’ and ‘Rab- 48’. These clones clearly differentiated rabies virus infected BHK- 21 cells with the mock infected cells using a cell-ELISA (Figuire 1).Hybridoma culture supernatant from clone ‘Rab-48’ was found to be relatively more efficacious in detection and titration of rabies virus in BHK-21 cells. Reactivity of a 2-fold dilution of this mAb with the rabies virus infected BHK-21 cells is shown in figure (Figure 2). This mAb was used extensively for generating data on growth kinetics of PV strain of rabies virus in BHK-21 cells.
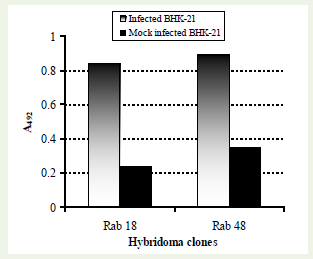
Figure 1: Reactivity of mAbs namely Rab-18 and Rab-48 in cell-ELISA. Hybridoma culture supernatants diluted 1:2 were screened against rabies virus infected BHK-21 cells and mock infected BHK-21 cells.
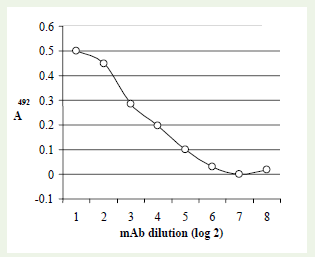
Figure 2: Reactivity of 2-fold dilution of mAb Rab-48 with rabies virus infected BHK-21 cells using a cell-ELISA.
The mAb based cell-ELISA was also used for the quantification of cell-free virus and cell-associated virus over a period of 9 days. The pattern of virus titre in ‘cell free’ and ‘cell associated’ samples indicated that the virus is internalized immediately and starts replicating. The cell free samples tested between 6 and 12 h post infection indicate a short eclipse phase, where the virus titre in the culture supernatant declines transiently. Subsequently, the virus quantities in ‘cell free’ sample as well as ‘cell associated’ samples follow an identical pattern with a maximum titre of the virus at around 48 h in both the samples. Subsequently the virus titre declines in the culture supernatant as well as within the cells. Virus titres in ‘cell-free’ and ‘cell-associated’ samples harvested at different time intervals is shown in the graph (Figure 3).
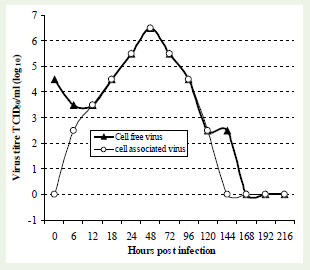
Figure 3: Rabies virus titration followed by cell-ELISA using microtitration method. Cell free and cell associated virus samples collected at different time intervals (0 to 9 days) were titrated. The cells were acetone fixed at 72h post infection and subjected to cell-ELISA.
mAb based cell-ELISA was used to study the growth pattern of rabies virus (PV strain) in BHK-21 cell system using adsorption as well as co-cultivation modes of cultivation. The co-cultivation method was found to be superior over virus adsorption with high virus yield.Therefore, for subsequent investigation of growth kinetics, cocultivation method was used. Although co-cultivation was superior for virus growth, the maximum virus titre was found at around 48 h of infection using both adsorptions as well as co-cultivation methods(Figure 4).
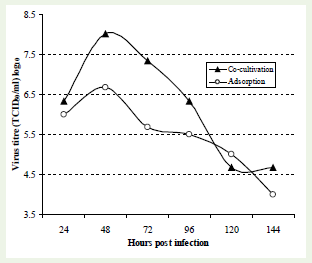
Figure 4: Growth kinetics of rabies virus by two different modes ofcultivation (co-cultivation & adsorption) using mAb based cell-ELISA. BHK-21 cells were infected with 0.1 MOI of rabies virus (PV strain) by cocultivation and adsorption (75% monolayer) mode of cultivation. Cell free virus was collected at 24 hrs intervals from day 1 (24 h) to day 6 (144 h). The collected samples were titrated by co-cultivation method using microtitration method and subjected to cell-ELISA using mAb ‘Rab-48’. The findings suggested that co-cultivation mode is superior to virus adsorption mode of cultivation with high virus yield.
FAT, Cell-ELISA and FACS analysis for assaying rabiesvirus growth kinetics
FAT using microtiration plates (micro-FAT) were performed on rabies virus infected cells using 10-fold dilution of virus samples harvested at various time intervals for the investigation of growth kinetics. Degree of fluorescence and FFU were observed under a fluorescent microscope. Distinct rabies virus specific intense cytoplasmic fluorescence were observed in positive samples. Amongst all the tests, FAT being universally accepted test for rabies virus detection was employed as a gold standard test.
Cell-ELISA was applied to investigate the growth pattern of PV strain of rabies virus in BHK-21 cells using virus at 1, 0.1 and 0.01 MOI. Virus titre in the samples collected using co-cultivation method at every 24 h intervals indicated that the inoculum with 0.1 MOI ofvirus is ideal for optimal virus/antigen preparation. This investigation indicated that the virus growth pattern using 0.1 and 0.01 MoI of virus inoculum were similar with an optimal harvest at around 48 h. The growth pattern with 1 MOI of virus inoculums showed relatively a flatgrowth curve. Findings suggested that optimum antigen production in BHK-21 cell system is possible by the use of 0.1 MOI of virus inoculums (Figure 5).
FACS technique was employed for the investigation of rabies virus growth kinetics in BHK-21 cells using rabies anti-nucleocapsid antibody conjugated with FITC. Pattern of distribution of cells with virus specific fluorescence in FACS analysis indicated an “eclipse” phenomenon of virus at around 12 h of infection, showing only 2.9 % infected cells. Subsequently, proportion of infected cells increased gradually showing 18.29% at 24 hand the maximum population of fluorescent cells were recorded at around 48 h (24.28%) of infection of virus. After 48 h, the proportion of cells showing fluorescence declined significantly to 5.3 % cells at 72 h Proportion of virus infected cells over a period of time using a single step growth curve is shown (Figure 6).
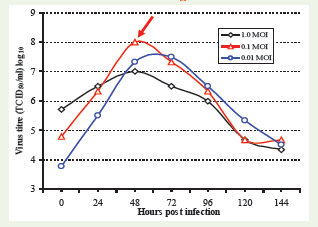
Figure 5: Growth pattern of rabies virus in BHK-21 cells using 1, 0.1and 0.01 MOI of virus by co-cultivation method. Note that 0.1 MOI of virus is most suitable for maximum virus titre, which is obtained at around 48 h post infection.
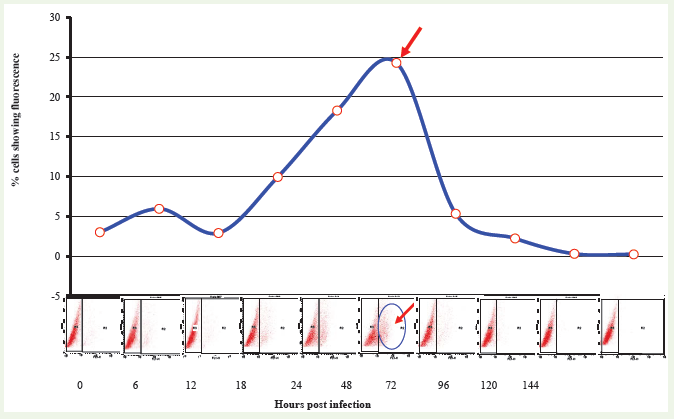
Figure 6: FACS analysis for investigation of growth kinetics of rabies virus. Cell free virus samples collected at different time intervals (0 h to 144 h) were titrated using 24 well culture plates. Cells infected at very low multiplity of virus were collected at 72 h post infection, acetone fixed and subjected to FACS analysis using rabies anti-nucleocapsid antibody conjugated with FITC. Point of the arrow indicates highest proportion of cells infected with virus at 48 h post infection.
Comparative efficacy of FAT, FACS analysis and cell-ELISA for the investigation of rabies virus growth kinetics
All the three tests employed in the present investigation were compared for their relative efficacy for the detection and titration of rabies virus in BHK-21 cell system. Micro-FAT indicated that the maximum virus titre was obtained at around 48 h, which again correlated well (r = 0.7509) with the findings in FACS analysis (Figure 7a). The efficacy of mAb based cell-ELISA was compared with micro- FAT for rabies virus quantification (r =0.8804). The cell-ELISA could detect the presence of rabies virus in BHK-21 cells when the virus foci were more than 3 per well as observed in identically processed samples. This finding explains that the cell-ELISA is relatively less sensitive compared to FAT for virus titration (Figure 7b). However, this test is suitable for deciding optimum time of harvest, the objective of this study.
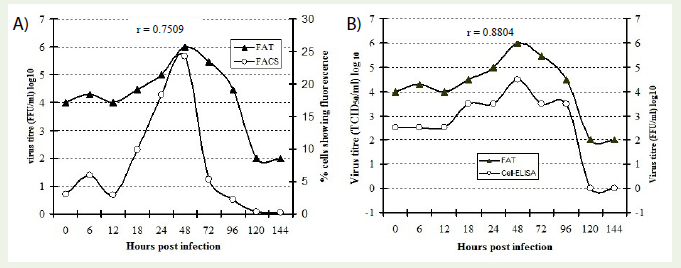
Figure 7: Comparative efficacy of FAT with that of FACS (7a) and cell-ELISA (7b) for rabies virus growth pattern in BHK-21 cells. Cell free virus samples collected at different time intervals (0 h to 144 h) were used for cell-ELISA, FAT and FACS.
Discussion
Rabies is a highly fatal viral zoonosis involving nervous system of warm-blooded animals including man. This disease under Indian socio-economic and socio-cultural conditions is spread mainly by the bite of rabid dogs. Major geographical region of India is classified as highly endemic [27]. Although, the disease is known to mankind since several centuries, sincere efforts to protect the livestock population from this deadly disease have not been given due importance. Vaccination of the animals following a pre-exposure or post exposure vaccination schedule is the only way to prevent rabies and to protect the animals from this deadly disease. The majority of the vaccines used in field at the moment are inactivated vaccines derived from cell culture. As is true for other inactivated vaccines like Foot and Mouth Disease (FMD), the quality and quantity of antigenic mass/antigenic value is of high significance. It is the main criteria for protective efficacy of such vaccines. Due to inadequate infrastructure for cell culture and lack of quality human resource in the developing countries, it is sometimes difficult to get desired antigenic value in the vaccine preparations. Therefore, in order to produce the vaccine with a desired antigenic value, cost of the per unit vaccine production increases resulting in a higher burden on people with low paying capacity. Under field conditions, an efficacious vaccine with a proven protective value/antigenic mass is must for use in large ruminants considering its economic value and risks after exposure to infection. Critical observations on the virus growth using certain in-process monitoring system is likely to reduce the production cost of the vaccine by simplification of the technology.
FAT for rabies virus quantification and rapid fluorescent foci inhibition test (RFFIT) for the assessment of post vaccinal immune response are considered to be the gold standard tests in rabies virus research [28,29]. Since rabies virus does not produce appreciable CPE in BHK-21 cells, virus titration is mainly dependent on expensive immunofluorescence or ethically concerned, cumbersome mouse inoculation tests. The protocols followed for the production of BPL inactivated vaccine is crippled by low virus yield of virus harvests that require frequent concentration steps and the lack of an userfriendly, economical assay for virus titration that can be performed on routine basis in a laboratory equipped with basic cell culture facilities. These tests require observation of individual samples/wells under the fluorescent microscope. On occasions owing to the poor staining techniques and poor quality of fluorescent microscopes FAT sometimes provides ambiguous results. Development of a cell- ELISA, which can provide quantitative assessment (digital values) of virus without the use of fluorescent microscopy, may be an added advantage due to more automation. This may also be useful for handling of large number of samples (virus and post vaccinal serum) during the launch of a rabies control programme.
Keeping all these problems in mind the present investigation aimed at finding out the optimal conditions for the harvest of the rabies virus using BHK-21 cells along with an objective of developing a homologous system for in-process monitoring of the quality of vaccine, so as to reduce the cost of vaccine production. The study involved propagation of rabies virus in BHK-21 cell system, production of mAb against rabies virus, development and application of cell-ELISA, FAT and FACS analysis for the investigation of growth kinetics of PV strain of rabies virus.
A polyclonal antibody based cell-ELISA was initially optimized for the detection/titration of rabies virus in BHK-21 cell system using microtitration method. As we know that polyclonal antibody based ELISAs give relatively inconsistent result owing to higher background absorbance (A492) values. Efforts to develop more precise and reproducible cell-ELISA were made by employing mAbs developed in the present study. The generation of mouse hybridoma clones against rabies virus antigen resulted in two useful clones for cell-ELISA, namely ‘Rab-18’ and ‘Rab-48’. Since the objective of developing these hybridoma clones was to use mainly in cell-ELISA, similar method involving rabies virus infected and mock infected BHK-21 cells were used for screening and selection of hybridoma clones. A similar strategy of screening of hybridoma clones that matches with the intended use of mAbs has been recommended earlier [222]. The mAb from the clone ‘Rab-48’ was used extensively for development and evaluation of cell-ELISA in growth kinetics of rabies virus. Although due to unavoidable circumstances these clones could not be saved. mAb based cell-ELISA was used to study the growth pattern of rabies virus in BHK-21 cell system. ‘Cell free’ as well as ‘cell associated’ virus samples were employed for the investigation of virus growth kinetics, following a methodology as reported earlier for peste des petits ruminants (PPR) virus [30]. Samples collected at different point of time, during the investigation indicated that the virus titre in the ‘cell free’ samples initially declines which is correlated with the increase in the virus quantity in ‘cell associated’ virus samples. The initial decline in the virus titre from ‘cell free’ samples between 6 to 12 h correspondto the “eclipse phase” of rabies virus [31,32] replication cycle. After 12 h the quantity of infectious virus (both cell free and cell associated virus) increased rapidly reaching to maximum at around 48 h after the exposure. Similar findings of rabies virus growth in BHK-21 cells have been reported earlier [18,33,34]. Virus growth pattern involving co-cultivation and adsorption modes of virus cultivation indicated that the former is superior to virus adsorption. The yield of infectious virus particles was significantly higher when inoculum was added at the time of cell seeding (co-cultivation) as compared to a preformed monolayer of BHK-21 cells.
It is a well established phenomenon that growth kinetics of a virus in any cell culture system is influenced by the quantity of the initial virus inoculum. Accordingly, for the construction of a single cycle growth curve 100 % of the cells have to be infected using 1 or more than one MOI of virus. The glycoprotein, which plays major role in the initial stage of infection, also happens to be major protective antigen of rabies virus [35]. The objective of the present investigation was to optimize the virus growth conditions for maximum infectious virus particles/glycoprotein antigen/protective antigen. Therefore, an experimental design involving different multiplicity of infection of virus ranging from 0.01, 0.1 and 1 MOI was developed. The findings with the ‘cell free’ virus samples collected up to 7 days post infection further suggested that the maximum infectious virus is obtained at around 48 h of infection. Therefore the collection of cell culture supernatant immediately after 48 h post infection may be more suitable for the preparation of vaccine. Previous studies have indicated that the yield of infectious rabies virus particles in BHK-21 cell suspension is directly proportional to the concentration of the cells [33]. The growth kinetics at low multiplicity of infection (0.1 and 0.01) suggest that the release of infectious virus particle from the infected cells shoots up to 48 h which also corresponds with the log phase of the growth of BHK-21 cells. Relatively a flat growth curve was observed at high multiplicity of infection (1.0 MOI). This could be possible because almost all the cells are infected immediately after infection. This may affect the growth rate of the cells thereby causing low virus titre throughout investigation.
FAT was applied as a gold standard test for monitoring of the growth pattern of rabies virus in BHK-21 cells as recommended [36]. For this purpose anti-nucleocapsid antibody conjugated to FITC (Bio- Rad, Cat.# 357-2114) was used as a tracing antibody. As we know that nucleocapsid (N) protein is a major internal virion protein involved in the T-cell activation [37], the level of fluorescence targeting N protein can be taken as a degree of virus replication. Number of FFU produced by a sample has extensively been used to establish correlation with mouse inoculation test in rabies virus research. Similarly the degree of inhibition of FFU (RFFIT) has extensively been studied and compared with the mouse protection test for the assessment of post vaccination immune response [28,38]. For determining the positivity of the sample, individual wells were examined under a fluorescent microscope and a well showing virus specific fluorescence even with a single focus was considered to be positive. A declining trend in the proportion of infected cells with specific fluorescence was observed with the decrease in virus inoculum (10-1 to 10-5) and practically no fluorescence in mock infected well (cell control).
The identical samples collected at various time intervals as in case of cell-ELISA were processed for FAT using a microtitration method.The comparative efficacy of FAT with that of cell-ELISA indicated that FAT is relatively more sensitive test than cell-ELISA. On an average mAb ‘Rab-48’ based cell-ELISA developed during the present study could detect a minimum of 3 to 10 FFU per well. This roughly indicates that the virus titres expressed in MAb based cell-ELISA are 1 to 2 log10 (10 to 100 times) lower than that in FAT using antinucleocapsid antibody conjugated with FITC. This is mainly owing to the two factors, 1) a positive detectable signal in mAb based ELISA may require a minimum population of infected cells however even single virus infected cell can be visualized in FAT. 2) FITC conjugated antibody although specific to rabies virus, is directed against several epitopes present in the nucleocapsid protein as compared to mAb Rab-48 against single epitope. Similar findings of the higher sensitivity of FAT have been observed when compared to mouse inoculation test previously [33].
In order to be more precise in determining the pattern of distribution of rabies virus infected cells at various time intervals, virus samples were analyzed by FACS analysis for growth kinetics. Findings of the FACS analysis represented the similar pattern of virus growth as methods cell-ELISA and FAT. Following the conventional virus titration methods on samples collected for FACS studies indicated a maximum proportion of the virus infected cells at around 48 h of virus exposure. The growth pattern of the virus showed a short eclipse phase at around 12 h followed by a steady increase in the proportion of infected cells up to 48 hrs. The titre of virus samples declined sharply after 48 h post infection.
The data on the mAb based cell-ELISA and FACS analysis shows potential application of these techniques for the investigation of growth kinetics of rabies virus. Although, all the three tests namely FAT, FACS analysis and cell-ELISA indicated that the maximum quantity of infectious virus particles could be harvested at around 48 h of inoculation with an inoculum quantity of 0.1 MOI, the antigenic value in terms of protective efficacy of vaccine needs to be established. The patterns of the results of the mAb based cell-ELISA shows thatit can replace the conventional FAT for the production and quality control of rabies vaccine. However, reproducibility of this test needs to be established. The study if applied successfully is likely to impact the production of the cell culture rabies vaccine, with the elimination of hurdles like concentration of virus and use of mice for vaccine virus titration, etc. Efforts are being made to take advantage of the findings of the present study in the rabies vaccine production and also to transfer the technology to various state veterinary biological units of India interested in rabies cell culture vaccine research and development and production.
Acknowledgements
Authors are great full to Joint Director, Academic and Director of Indian Veterinary Research Institute for the support. This research work was in part funded under the Project “Niche area of Excellence on Vaccines and Diagnostics”.
References
- Van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB (2000). Family rhabdoviridae. In: proceedings of the seventh report of the international committee on taxonomy. Classification and nomenclature of viruses: Virus taxonomy, Classification and nomenclature of viruses. New York Academic press: 563-583.
- WHO (1973) Sixth report of the Expert committee on Rabies. Technical report series, 523. World Health Organization, Geneva, Switzerland.
- Fischman H, Ward FE (1968) Oral transmission of rabies virus in experimental animals. Am J Epidem 88: 132-138.
- Constantine DG (1967) Rabies virus transmission by air in bat caves. U.S. public health service, Washington DC.
- Haupt W (1999) Rabies-risk of exposure and current trends in prevention of human cases. Vaccine 17: 1742-1749.
- Wilde H, Tipkong P, Khawplod P (1999) Economic issues in postexposure rabies treatment. J Travel Med 6: 238-242.
- Meslin FX, Fishbein DB, Matter HC (1994) Rationale and prospects for rabies elimination in developing countries. In C.E. Rupprecht, B. Dietzschold, and H. Koprowski (ed.), Lyssaviruses. Springer-Verlag KG, Berlin, Germany: 1-26.
- Rupprecht CE, Smith JS, Fekadu M, Childs JE (1995) The ascension of wildlife rabies: a cause for public health concern or intervention? Emerg Infect Disn 1: 107-114.
- Kawai A, Morimoto K (1994) Functional aspects of lyssavirus proteins, In: CE. Rupprecht, B. Dietzschold and H. Koprowski (ed.), Lyssaviruses. Springer-Verlag KG, Berlin, Germany: 27-42.
- Bektemirova MS, Osidze DF, Pille ER, Nadaichik LV, Matevosyan KS et al. (1979) Properties of Rabies virus (MNIIVP-74 strain) adapted to Japanese quail embryo cell culture. Arch Virol 61: 61-68.
- Tierkel ES, Atanasiu P (1996) Rapid microscopic examination for Negri bodies and preparation of specimens for biological tests. In: Meslin, F.X., Kaplan, MM. and Koprowski, H. (ed.). Laboratory techniques in rabies. 4th ed. WHO, Geneva: 55-62.
- Goldwasser RA, Kissling RE (1958) Fluorescent antibody staining of street and fixed rabies virus antigens. Proc Soc Exp Bio Med 98: 219-223.
- Koprowski H (1973) The mouse inoculation test. In: Meslin FX, Kaplan MM, Koprowski H, (ed.). Laboratory techniques in rabies. 3th ed. WHO, Geneva: 85-93.
- King AA (1996) Cell culture of rabies virus. In: Meslin, F.X., Kaplan, M.M. and Koprowski, H. (ed.). Laboratory techniques in rabies. 4th ed. WHO, Geneva: 114-130.
- Perrin P, Rollin PE, Sureau PA (1986) A rapid rabies enzyme immunodiagnosis (RREID): a useful and simple technique for the routine diagnosis of rabies. J Biol Stand 14: 217-222.
- Wiktor TJ, Flamand A, Koprowski H (1980) Use of monoclonal antibodies in diagnosis of rabies virus infections and differentiation of rabies and rabies-related viruses. J Virol Methods 1: 33-46.
- Hummeler K, Atanasiu P (1996) Electron Microscopy. In: Meslin, F.X., Kaplan, M.M. and Koprowski, H., (Eds). Laboratory techniques in rabies. 4th ed. WHO, Geneva: 85-93.
- Bordignon J, Ferrira SCP, Caporale GMM, Carrieri ML, Kotait I, et al. (2002) Flowcytometry assay for intracellular rabies virus detection. J Virol Methods 105: 181-186.
- Sharma RN (1990) Development of inactivated Rabies cell culture vaccine. Ind J Virol 6: 83-86.
- Sarkar J, Belaineh G, Sreenivasa BP, Singh RP (2012) Development of a cell-ELISA using anti-nucleocapsid protein monoclonal antibody for the titration of PPR vaccine virus. Indian J Comp Microbiol Immunol Infect Dis 33: 18-20.
- Samuel D, Megson B, Strang M, Appleton H (2000) A micro titer plate method for titration and typing of poliovirus using a blue-cell ELISA. J Virol Methods 90: 125-133.
- Singh RP, Bandyopadhyay SK, Sreenivasa BP, Dhar P (2004) Production and Characterization of Monoclonal Antibodies to Peste des Petits Ruminants (PPR) Virus. Vet Res Comm 28: 623-639.
- Singh RP (2002) Production and characterization of monoclonal antibodies to peste des petits ruminants (PPR) virus. (Ph.D thesis, Deemed University, Indian Veterinary Research Institute, Izatnagar).
- Harlow E, Lane D (1988) Antibodies: a laboratory manual. Cold Spring Harbour Laboratory, New York :148-242.
- Peters JH, Baumgarten H (1992) Monoclonal Antibodies. Springer Laboratory: 51-390.
- Reed LJ, Muench H (1938) A simple method of estimating fifty per cent endpoints. Am J Hyg 27: 493-497.
- WHO (2001) Epidemiology of rabies.
- Zalan E, Wilson C, Pukitis D (1979) A microtest for the quantitation of rabies virus neutralizing antibodies. J Biol Stand 7: 213.
- Cho HC, Fenje P (1975) Rabies neutralizing antibody determination in tissue culture by direct fluorescent antibody technique. J Biol Stand 3: 101-105.
- Sreenivasa BP, Singh RP, Mondal B, Dhar P, Bandyopadhyay SK (2006) Marmoset B95a cells: A sensitive system for cultivation of Peste des Petits Ruminants (PPR) Virus. Vet Res Comm 30: 103-108.
- Matsumoto S (1974) Morphology of rabies virion and cytopathology of virus infected cells. Symp Ser Immunobiol Handb 21: 25-34.
- Iwasaki Y, Wiktor TJ, Koprowski H (1973) Early events of rabies virus replication in tissue cultures. An electron microscopic study. Lab Invest 28: 142-148.
- Chapman WG, Ramshaw IA, Crick J (1973) Inactivated rabies vaccine produced from the Flury LEP strain of virus grown in BHK-21 suspension cells. App Microbiol 26: 858-862.
- Kaplan MM, Wicktor TJ, Maes RF, Campbell JB, Koprowski H (1967) Effect of polyions on the infectivity of rabies virus in tissue culture: construction of a single-cycle growth curve. J Virology 1: 145-151.
- Cox JH, Dietzschold B, Schneider LG (1977) Rabies virus glycoprotein II. Biological and serological characterization. Infect Immun 16: 754-759.
- Dean DJ, Abelseth MK (1973) The fluorescent antibody test. In: M.M. Kaplan and H. Koprowski (eds). Laboratory techniques in rabies. 3rd ed. WHO, Geneva: 73-83.
- Celis E, Karr RW, Dietzschold B, Wunner WH, Koprowski H (1988a) Genetic restriction and fine specificity of human T-cell clones reactive with rabies virus. J Immunol 141: 2721.
- Smith JS, Yager PA, Baer GM (1973) A rapid reproducible test for determining rabies neutralizing antibody. Bull WHO 5: 902.