Review Article
DNA Metabarcoding: A New Approach forRapid Biodiversity Assessment
Pavan-Kumar A*, Gireesh-Babu P and Lakra WS
Corresponding author: Dr. Annam Pavan Kumar, Scientist, Division of Fish Genetics and Biotechnology, ICAR-CentralInstitute of Fisheries Education, Versova, Mumbai-61, India,; E-mail: pavanannam@gmail.com
Citation: Pavan-Kumar A, Gireesh-Babu P, Lakra WS. DNA Metabarcoding: A New Approach for Rapid Biodiversity Assessment. J Cell Sci Molecul02 Biol. 2015;2(1): 111.
Copyright © 2015 Pavan-Kumar A et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Submission: 29/11/2014; Accepted: 25/02/2015; Published: 06/03/2015
Abstract
Biodiversity characterization is important to understand the ecological processes on earth. The recent advancements in molecular techniques have enabled us to identify the species composition more efficiently than the traditional methods. In DNA metabarcoding, the pooled genomic DNA extracted from environmental samples is used to amplify evolutionarily conserved genes by universal primers and sequenced using next generation sequencing technologies. In this brief review, the concept of DNA metabarcoding and its applications, limitations and challenges have been discussed.
Keywords: Biodiversity; DNA metabarcoding; Next Generation Sequencing; Environmental DNA
Introduction
The extant /present biodiversity is a result of several million years of evolution of life on earth. Biodiversity is a key component of ecosystem and plays a major role in proper functioning of the ecosystem. Several factors like climate change, habitat loss and invasive species are disturbing the ecosystem biotic components thereby adversely affecting the function and services of ecosystem [1]. The effective management measures for restoring the degraded ecosystem can be taken if the information (data) about indicator species / abundance and pattern of biological diversity (Species) in that ecosystem is available. Traditional morphological and meristic tools for characterizing and assessing the biodiversity demands high skilled personnel and have limitations in identifying cryptic species. With the advent of molecular biology, DNA based species identification methods have been devised using molecular markers(mitochondrial and nuclear). Since the last decade, taxonomically informative genes have been tested over large groups of organisms(animals: mitochondrial cytochrome c oxidase subunit I [2]; Fungi: Nuclear ribosomal internal transcribed spacer [3]; Plants: two chloroplast genes, rbcL & matK [4]; Bacteria: 16S rRNA & protein coding Chaperonin-60, cpn60 [5-6] for their efficiency to delimit the species and designated them as barcode genes for respective groups. The success in this approach resulted in creation of huge reference databases that include species taxonomic details along with DNA barcode gene sequences(Fish-BOL, BOLD, MarBOL, QBOL etc). These reference barcode sequence databases are useful in assigning taxon to unknown specimen by comparing the sequence similarity of specimen barcode gene with reference database. Until recently, most of the barcoding studies were aimed at developing reference databases by generating species specific DNA barcodes from individual specimens. However, it is also important to characterize/assess the species diversity and abundance within an ecosystem as a whole to understand the spatial and temporal changes in species diversity [7]. Normal DNA barcoding approach using Sanger sequencing method can identify only one specimen at a time and cannot identify multiple species if the sample contains a mixture of different species. With the advancements in sequencing technology, it is now possible to assess the species composition of ecosystems including environmental samples such as soil, sediment and water at a stretch than screening individual specimens at a time.
DNA Metabarcoding
Taberlet et al. [8] introduced the term DNA metabarcoding todesignate high-throughput multispecies identification using the totalor typically degraded DNA extracted from an environment sample orfrom bulk samples of entire organisms. The multispecies identificationtechnique was originally applied to microbial communities [9] in thename of metagenomics, and now it is being applied for eukaryotic organisms such as fungi [10], invertebrates [11], plants [12] andvertebrates [13-15]. Metabarcoding differs from metagenomics inseveral ways as metagenomics refers to the study of all genomes withina particular ecosystem whereas metabarcoding aims to study a subsetof genes / gene. From methodology point of view, metagenomicsapproach includes preparation of shotgun (random) libraries forsequencing while metabarcoding is based on amplicon sequencing.Metagenomics approach generally used to get more insights aboutthe interaction between species within an ecosystem (taxonomic andfunctional information). Metabarcoding approach is mainly used todocument / characterize species diversity in the ecosystem and it canhave better coverage to identify rare taxa within an ecosystem.
DNA Metabarcoding methodology
Selection of Next Generation Sequencing (NGS) technology
A series of high-throughput sequencing technologies based ondifferent chemistries and detection techniques have been introducedcommercially. All these NGS technologies can generate severalhundred thousands of millions of sequencing reads in parallel. Thismassively parallel throughput sequencing capacity can generatesequence reads from fragmented libraries of a specific genome (i.e.genome sequencing) or from a pool of PCR amplified molecules(i.e. amplicon sequencing, [16]). Metabarcoding approach relieson this technology where large number of amplicons of taxonomicinformative (barcode) gene can be sequenced without a necessity forcloning [8]. A comparison of currently available NGS platforms isgiven in Table 1. Till now, most of the DNA metabarcoding studieshave used Roche 454 FLX platform due to its ability to produce longread length and relatively short run time. However, this platformcannot read homopolymers accurately and may provide erroneoussequences. This problem has been largely alleviated by implyingbioinformtic tools that filter out erroneous sequences [17]. Othersequencing technologies such as Illumina, SoLiD and Ion torrentplatforms can read homopolymers accurately but the read lengthis relatively low. Selection of appropriate sequencing methodologydepends on the question to be addressed and length of fragment(amplicon). If amplicon length is short (100-200bp), Illumina andIon torrent platforms are appropriate whereas for large ampliconsRoche GS FLX is more useful.
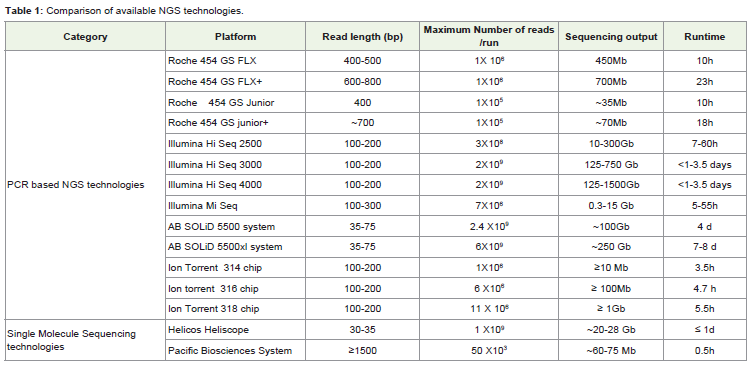
Table 1: Comparison of available NGS technologies.
PCR amplification of DNA
A certain genomic region can be amplified from the DNAextracted from sample that contains a mixture of species. Althoughany part of the genome can be used to delimit the species, certainfeatures like mutation rate (molecular evolution rate), availability ofuniversal primers, short sequences with sufficient phylogenetic signaland availability of comprehensive taxonomic reference database aresome of the important features to consider before selecting a DNAfragment for metabarcoding studies. Some researchers have usedshort fragments of the nuclear 18S and 28S ribosomal markers formetazoans [18-19], but these regions may underestimate speciesdiversity due to their slow rate of evolution compared to othermitochondrial markers [20-22]. Short fragment of mitochondrial 12Sribosomal gene has successfully been used for delineating metazoans,however, taxonomic reference databases are limited for this marker compared to cytochrome c oxidase subunit I [18]. The mitochondrialpartial cytochrome c oxidase I gene (COI) has been adopted asthe standard barcode gene for most of the animal groups [3] andthis gene has the most represented taxonomic reference databasein public domain. However, through in silico [23] and empiricalanalyses it has been found that the available universal primers forCOI gene are not well conserved in certain groups viz., nematodes[22,24], echinoderms [25] and gastropods [26]. In summary, theaccuracy of metabarcoding is highly dependent on marker choice, butunfortunately no marker has all the features to be used as a perfectmetabarcoding marker and the best marker choice could be studyspecific[27]. Ficetola et al. [23] have developed a software ecoPCR(electronic PCR) to test the efficacy of barcode primers. Based on twoparameters: barcode coverage (Bc) and barcode specificity (Bs), thismethod measures the conservation of the primers and the capacity ofthe amplified region to discriminate between taxa [28]. This softwarefacilitates a preliminary comparison of several DNA regions toidentify the most appropriate barcodes. A summary of the genes /markers used for various metabarcoding studies is given in Table 2.
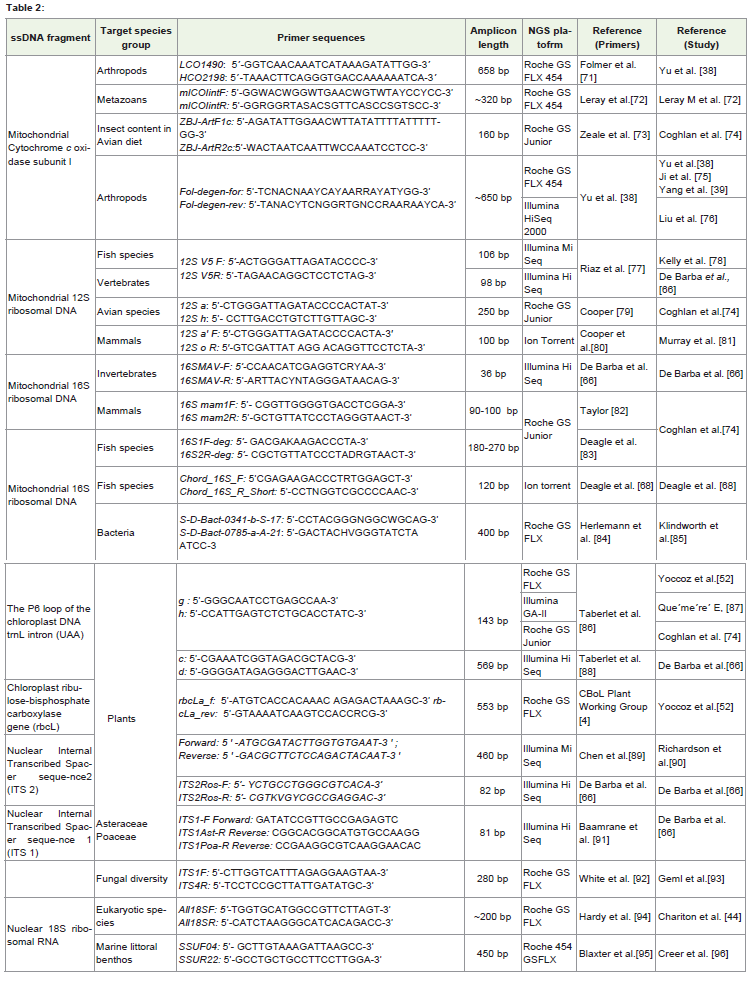
Table 2:
Amplicon multiplexing, library preparation andsequencing
Generally, NGS platforms are intended to sequence wholegenome of important model and non-model organisms and duringthis process they generate thousands of millions of reads perinvestigation. However, metabarcoding studies aims to sequence shortfragment of homologous gene (amplicon sequencing) from differentspecies of many samples. Sequencing each sample (contains manyorganisms) separately is not economical. Multiplexing of sampleswith short, identifying sequences (barcodes/ multiplex identifier(MID)/index) is a widely used strategy in which different moleculartags (4-5 nucleotides) are attached to all DNA fragments / ampliconsto identify samples. This kind of sequence indexing is used for datasorting after sequencing and to assign the sequence reads to specificsamples. The most effective way to produce an indexed ampliconsis to amplify the genomic target region using PCR with specificprimers that include a sequencing adaptor and a barcode (Figure 1) [29-30]. Other indexing strategies rely on ligation of barcodes orbarcoded sequencing adapters to the DNA amplicons [31]. Afterindexing, amplicons can be pooled (equimolar concentration of eachamplicon) and sequencing would be carried out using the pooledbarcoded libraries. The number of different samples to be pooledper sequencing run is determined by number of barcodes available.High quality reagents with barcoded adapters and PCR primers arereadily available in kits from many vendors. After tagging with MIDs,the amplicon library fragments are clonally amplified onto the beadparticles through emulsion PCR. These particles containing sequenceclones are deposited on chips/ flow cells for sequencing by NGSplatforms. Designing of tags / MID sequences is important as thesesequences may lead to PCR bias and several software are availablefor this purpose (BARCRAWL [32]; oligotag program of OBITools,http://www.gren oble.prabi.fr/trac/OBITools).
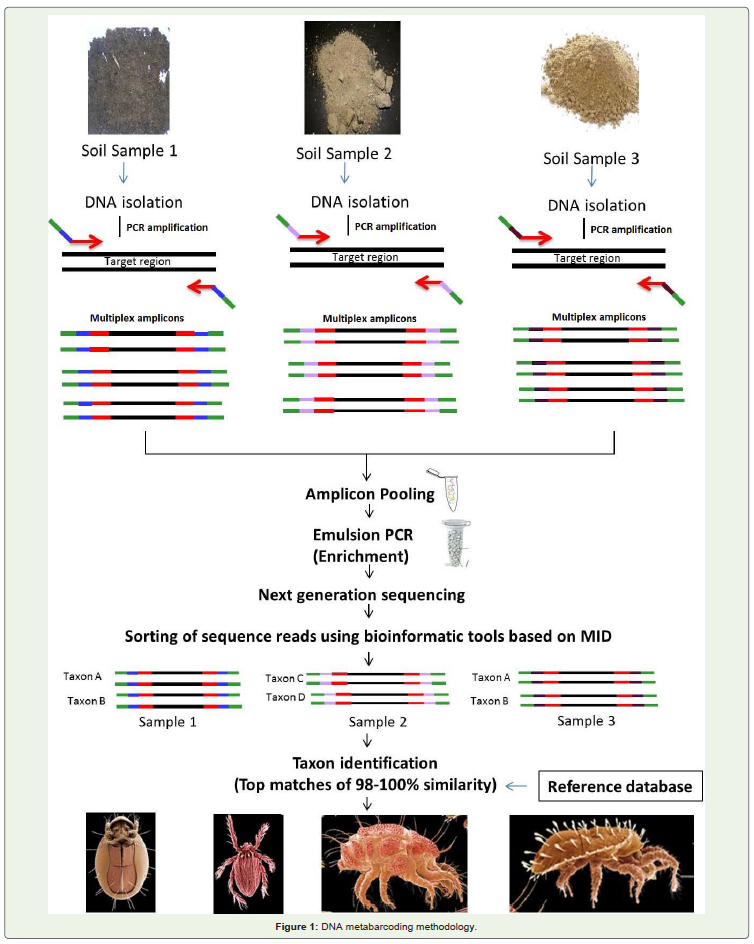
Figure 1: DNA metabarcoding methodology.
Data analysis
The data generated by NGS platforms are huge and traditionalcomputer operating systems (Windows/DOS) are not capable to handle the data. UNIX operating system has been considered as thestandard computing environment for NGS data. Further, most ofthe bioinformatics software programs/ algorithms are compatiblewith UNIX operating system. In UNIX, tasks can be performed bywriting commands and in personal computers UNIX environmentcan be provided by installing Linux. Certain operating systems suchMac OSX (Apple Inc) provide UNIX environment that uses bothGraphical User Interfaces (GUI) and command mode.
NGS output data consists of DNA sequences (reads) andcorresponding quality values (for each nucleotide of each sequenceread). All the resulting sequences may not represent the speciescomposition of sample. The sequence data may contain sequencingnoise, PCR chimeras [33], contaminant sequences, nuclearmitochondrial pseudogenes (Numts) and PCR errors. Eliminating allthese error sequences from final data analysis is prerequisite beforeassigning taxon to the sequences. In brief, the data analysis consists ofthree steps viz., data pre-processing (removal of primers, sequencingadaptors and demultiplexing), processing raw reads (denoising,chimera and PCR artefacts removal) and performing analyses(clustering, BLASTing) (Figure 2). Different software / algorithmshave been developed to perform the above tasks. Different researchershave designed different pipelines by combining various software toanalyse NGS metabarcode data. Among different packages, QIIME(Quantitative Insights Into Microbial Ecology [341]) has beensuccessfully used for different metabarcoding and metageneticsstudies [35-37]. QIIME is an open-source bioinformatics pipelineconsists of native python code and additionally cover many externalapplications for data analysis from raw data processing to taxonomicassignment. It is initially developed for microbial community however; QIIME environment is now being used for metazoan andplant metabarcoding studies [38,39].
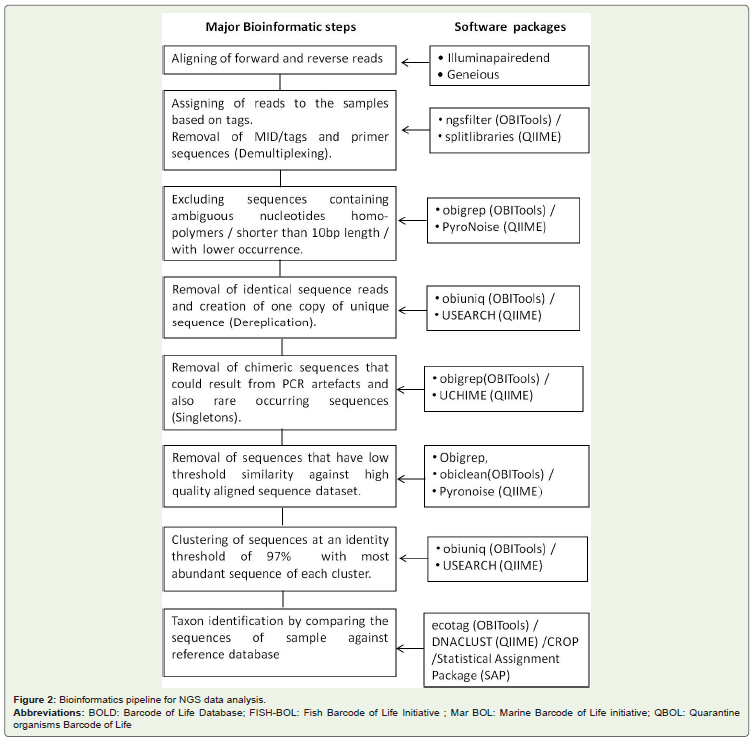
Figure 2: Bioinformatics pipeline for NGS data analysis. Abbreviations: BOLD: Barcode of Life Database; FISH-BOL: Fish Barcode of Life Initiative ; Mar BOL: Marine Barcode of Life initiative; QBOL: Quarantineorganisms Barcode of Life
Likewise, OBITools is another open source package that has beenspecifically designed for analyzing metabarcoding data. The mainadvantage of the OBITools is their ability to take into account thetaxonomic annotations, ultimately allowing sorting and filtering ofsequence records based on the taxonomy. Apart from these software,some other packages like Operational Clustering of Taxonomic Unitsfrom Parallel UltraSequencing (OCTUPUS) Bioconductor packagesShortRead [40] and Biostrings (run on R language) are also availablefor NGS data analysis.
In metabarcoding, species are defined operationally as a clusterof similar sequences, and the clusters are known as OperationalTaxonomic Unit (OTU). The most critical step in the analysis is toassign taxon to sequences / cluster of sequences (OTU). This can beachieved by comparing each sequence to a reference database that isa subset of public databases (eg. EMBL, NCBI GenBank, SILVIA andBOLD) or a set of sequences specifically produced for the study. Thecomparison for sequence similarity between queried sequence andreference database sequence can be performed through BLAST searchor ecotage [41]. In the case of non availability of reference database,sequences would not be linked to a taxonomic name, however; wouldbe clustered in MOTUs (Molecular Operational Taxonomic units)that can be compared in different studies, for example, comparingthe diversity of MOTUs in different localities or under differentparameters in the same locality [28]. The Program MEGAN (MEtaGenome Analyzer) is useful in representing the species/ taxoncomposition of sample and for taxonomic binning, even for verylarge data sets [42].
Data analysis
Biodiversity studies
In this decade of biodiversity (2011-2020), fund allocation has beenincreased for biodiversity characterization and these efforts resultedin creation / strengthening of existing taxonomic reference databasessuch as Global Biodiversity Information Facility (GBIF) and Barcodeof Life database (BOLD). These databases have cured referencetaxonomic information for about 144,357 Species (BOLD www.boldsystems.org.in [43]) species and are being constantly updatingwith new taxon information to include all the species on earth. Once the comprehensive database is prepared, DNA metabarcodingapproach can be used to analyse the species composition of differenttypes of samples, from soils to sediments, faeces, air and water.Metabarcoding of soil / sediment samples collected from nuclearpower plant areas or any other industrial area can be used to comparetemporal and spatial species assemblages to assess human impactson biodiversity [44]. Likewise, water samples can be used to detectthe presence of invasive species [45]. Next generation sequencingtechnology has been used to analyse species composition ofsensitive ecosystem (Coral reefs [46]), extreme habitats (acid mines[47]). DNA metabarcoding approach has been successfully used to characterize soil microbial diversity [48-50], fungal diversity [51] andplant diversity [52] using 16S rRNA, ITS and P6 loop of the plastidDNA trnL intron amplicons, respectively. Hajibabaei et al. [53] wereused short fragments of COI DNA barcodes were used to identifyfreshwater macro invertebrates from benthic samples.
The effect of climate change on biodiversity could be assessedand species distributions can be predicted for future if data on pastdistributions together with past climate conditions are available.DNA metabarcoding of soil samples collected at different depthscould provide a new source of information about past speciesdistributions [7]. Murray et al. [54] have analysed ancient DNAusing metabarcoding approach and identified diverse range of taxa,including endemic, extirpated and previously unrecorded taxa.Haouchar et al. [55] assessed ancient DNA of vertebrate fossils andplants and provided valuable information about past biodiversityof Kangaroo Island, Australia. Haile et al. [56] utilized both 454pyrosequencing and conventional Sanger sequencing methods in theanalysis of ancient DNA recovered from Arctic permafrost cores.
Trophic studies
The interaction between predator and prey play an importantrole in maintaining ecosystem health and stability. Gut contentanalysis of the species to identify their feeding habits, especially forendangered species will help to formulate conservation measures/strategies [57]. Traditionally, the diet composition of any species isassessed by macro- or micro histological methods [58] and stableisotopes [59]. However, these methods are time consuming, requirehighly skilled personnel and cannot identify variably digested fooditems. To overcome this, DNA isolated from gut content and faecescan be used for the molecular identification of diet compositionby metabarcoding. Several studies have used NGS technology forinvestigating gut microbe ecology and species composition of diet.Some of these studies have included analyses of herbivore diet fromgut contents using the plastid trnL sequence [13, 41, 60-61]. Severalstudies investigated the species composition of diet by analysingprey DNA collected from faeces of Australian fur seal (Arctocephaluspusillus doriferus [27]) little penguin (Eudyptula minor [62-63]),reptile [15] leopard cat [64]. Recently De Barba et al., [66] analysedplant, vertebrate and invertebrate components of the diet of brownbear by analysing faecal matter through multiplexing strategy.
Limitations
Metabarcoding, like many new technological advances in science,offers new opportunities and at the same time new challenges. Sincemetabarcoding studies generally include amplicon sequencing,factors like PCR efficiency, primer tags and sequencing efficacy needto be considered to avoid errors [67, 68, 69]. To circumvent primertag bias, a two stage PCR where template DNA is first amplified usinguntagged primers and subsequently by tagged primers during the lastfew PCR cycles has been suggested [53,68]. Another limitation is lackof comprehensively cured reference databases for certain metazoansfor assigning taxon to the OTUS. Future studies are needed toimprove sampling strategies (selection of season, sampling locationwithin habitat) and to understand the relationship between sequencereads and species density [70]. Further, the integration of knowledge from ecology, taxonomy and evolution is essential for addressing anybiodiversity questions using metabarcoding.
References
- Sala OE, Chapin FS, Armesto JJ, Berlow E, Bloomfield J, et al. (2000) Global biodiversity scenarios for the year 2100. Science 287: 1770-1774.
- Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc Biol Sci 270: 313-322.
- Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, et al. (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109: 6241-6246.
- CBoL Plant Working Group (2009) A DNA barcode for land plants. Proc Natl Acad Sci USA 106: 12794-12797.
- Makarova O, Contaldo N, Paltrinieri S, Kawube G, Bertaccini A, Nicolaisen M (2012) DNA barcoding for identification of “Candidatus phytoplasmas†using a fragment of the elongation factor Tu gene, PLoS ONE.
- Links MG, Dumonceaux TJ, Hemmingsen SM, Hill JE (2012) The chaperonin-60 universal target is a barcode for bacteria that enables de novo assembly of metagenomic sequence data. PLoS One.
- Yoccoz GN (2012a) The future of environmental DNA in ecology. Mol Ecol 21: 2031-2038.
- Taberlet P, Coissac E, Pompanon F, Brochmann C, Willerslev E (2012) Towards next-generation biodiversity assessment using DNA metabarcoding. Mol Ecol 21: 2045-2050.
- Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, et al. (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere.â€. Proc Natl Acad Sci USA 103: 12115-12120.
- Fouts DE, Szpakowski S, Purushe J, Torralba M, Waterman RC, et al. (2012) Next Generation Sequencing to Define Prokaryotic and Fungal Diversity in the Bovine Rumen. PLoS One 7: e48289.
- Porazinska DL, Giblin-Davis RM, Esquivel A, Powers TO, Sung W, et al. (2010) Ecometagenetics confirms high tropical nematode diversity. Mol Ecol 19: 5521-5530.
- Hiiesalu I, Opik M, Metsis M, Lilje L, Davison J, et al. (2012) Plant species richness belowground: higher richness and new patterns revealed by next-generation sequencing. Mol Ecol 21: 2004-2016.
- Kowalczyk R, Taberlet P, Coissac E, Valentini A, Mique C et al. (2011) Influence of management practices on large herbivore diet—Case of European bison in Białowieza Primeval Forest (Poland). F Ecology and Management 261: 821-828.
- Raye´ G, Miquel C, Coissac E, Redjadj C, Loison A, et al. (2011) New insights on diet variability revealed by DNA barcoding and high-throughput pyrosequencing: chamois diet in autumn as a case study. Eco Res 26: 265-276.
- Brown DS, Jarman SN, Symondson WO (2012) Pyrosequencing of prey DNA in reptile faeces: analysis of earthworm consumption by slow worms. Mol Ecol Res 12: 259-266.
- Shokralla S, Spall JL, Gibson JF, Hajibabaei M (2012) Next-generation sequencing technologies for environmental DNA research. Mol Ecol 21: 1794-1805.
- Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, et al. (2009) Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods 6: 639-641.
- Machida RJ, Knowlton N (2012) PCR Primers for metazoan nuclear 18S and 28S ribosomal DNA sequences. PLoS One, 7:e46180.
- Fonseca VG, Carvalho GR, Sung W, Johnson HF, Power DM, et al. (2011) Second-generation environmental sequencing unmasks marine metazoan biodiversity. Nat Commun 1: 98 doi:10.1038/ncomms1095.
- Hillis D, Dixon M (1991) Ribosomal DNA - molecular evolution and phylogenetic inference. Q Rev Biol 66: 411-453.
- Tautz D, Arctander P, Minelli A, Thomas RH, Vogler AP (2003) A plea for DNA taxonomy. Trends Ecol Evol 18: 70-74.
- Derycke S, Vanaverbeke J, Rigaux A, Backeljau T, Moens T (2010) Exploring the use of Cytochrome Oxidase c Subunit 1 (COI) for DNA barcoding of free living marine nematodes. PLoS One 5: e13716.
- Ficetola GF, Coissac E, Zundel S, Riaz T, Shehzad W, et al. (2010) An in silico approach for the evaluation of DNA barcodes. BMC Genomics 11: e434 doi: 10.1186/1471-2164-11-434.
- Creer S, Fonseca VG, Porazinska DL, Giblin-Davis RM, Sung W, et al. (2011) Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Mol Ecol 19: 4-20.
- Hoareau TB, Boissin E (2010) Design of phylum-specific hybrid primers for DNA barcoding: addressing the need for efficient COI amplification in the Echinodermata. Mol Ecol Res 10: 960-967.
- Meyer CP, Paulay G (2005) DNA Barcoding: Error Rates Based on Comprehensive Sampling. Godfray C, ed. PLoS Biology 3:e422.
- Deagle BE, Kirkwood R, Jarman SN (2009) Analysis of Australian fur seal diet by pyrosequencing prey DNA in faeces. Mol Ecol 18: 2022-2038.
- Coissac E (2012) OligoTag a program for designing set of tags for next generation sequencing of multiplexed samples. In: Data Production and Analysis in Population Genomics (eds Bonin A and Pompanon F). Methods in Molecular Biology Series, Humana Press, New York.
- Binladen J, Gilbert MTP, Bollback JP, Panitz F, Bendixen C, Nielsen R, Willerslev E (2007) The use of coded PCR primers enables high throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One 2:e197.
- Hoffmann C, Minkah N, Leipzig J, Wang G, Arens MQ, et al. (2007). DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res 35:e91.
- Meyer M, Stenzel U, Hofreiter M (2008) Parallel tagged sequencing on the 454 platform. Nat Protoc 3: 267-278.
- Frank DN (2009) BARCRAWL and BARTAB: software tools for the design and implementation of barcoded primers for highly multiplexed DNA sequencing. BMC Bioinformatics 10: 362.
- Lenz T, Becker, S (2008) Simple approach to reduce PCR artefact formation leads to reliable genotyping of MHC and other highly polymorphic loci - implications for evolutionary analysis. Gene 427: 117-122.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) Qiime allows analysis of high-throughput community sequencing data. Nature Meth 7: 335-336.
- Brinkman BM, Hildebrand F, Kubica M, Goosens D, Favero JD, et al. (2011) Caspase deficiency alters the murine gut microbiome. Cell Death and Dis 2: e220.
- Flores GE, Bates ST, Knights D, Lauber CL, Stombaugh J, et al. (2011) Microbial biogeography of public restroom surfaces. PLoS One 6: e28132.
- Kostka JE, Prakash O, Overholt WA, Green SJ, Freyer G, et al. (2011) Hydrocarbon degrading bacteria and the bacterial community response in gulf of mexico beach sands impacted by the deepwater horizon oil spill. Appl Environ Microbiol 77: 7962-7974.
- Yu DW, Ji YQ, Emerson BC, Wang XY, Ye CX, et al. (2012) Biodiversity Soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol Evol 3: 613-623.
- Yang C, Wang X, Miller JA, Blécourt M, Ji Y, et al. (2014) Using metabarcoding to ask if easily collected soil and leaf-litter samples can be used as a general biodiversity indicator. Ecol Indic 46: 379-389.
- Morgan M, Anders S, Lawrence M, Aboyoun P, Pages H, et al. (2009) ShortRead: a Bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25: 2607-2608.
- Pegard A, Miquel C, Valentini A, coissac E, Bouvier F, et al. (2009) Universal DNA-based methods for assessing the diet of grazing livestock and wildlife from faeces. J Agric Food Chem 57: 5700-5706.
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21: 1552-1560.
- Ratnasingham S, Hebert PDN (2007) BOLD: The Barcode of Life Data System. Mol Ecol Notes 7: 355-364.
- Chariton AA, Court LN, Hartley DM, Coloff MJ, Hardy CM (2010) Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Front in Ecol and Envi 8: 233-238.
- Ficetola GF, Miaud C, Pompanon F, Taberlet P (2008) Species detection using environmental DNA from water samples. Biol Lett 4: 423-425.
- Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F (2007) Metagenomic analysis of the microbial community associated with the coral Poritesastreoides. Envi Micro 9: 2707-2719.
- Edwards RA, Rodriguez-Brito B, Wegley L, Matthew Haynes, Mya Breitbart, et al. (2006) Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genom 7: 57.
- Roesch LFW, Fulthorpe RR, Riva A, Casella G, Hadwin AK, et al. (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. The ISME Journal 1: 283-290.
- Rousk J, Baath E, Brookes PC, Lauber CL, Lozupone C, et al. (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4: 1340-1351.
- Jumpponen A, Jones KL, Blair J (2010) Vertical distribution of fungal communities in tall grass prairie soil. Mycologia 102: 1027-1041.
- Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, et al. (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere.â€. Proc Natl Acad Sci USA 103: 12115-12120.
- Yoccoz NG, Brathen KA, Gielly L, Haile J, Edwards ME et al. (2012b) DNA from soil mirrors plant taxonomic and growth form diversity. Mol Ecol 21: 3647-3655.
- Hajibabaei M, Shokralla S, Zhou X, Singer GAC, Baird DJ (2011) Environmental barcoding: a next-generation sequencing approach for biomonitoring applications using river benthos. PLoS ONE 6: e17497.
- Murray DC, Haile J, Dortch J, White NE, Haouchar D, et al. (2013) Scrapheap Challenge: A novel bulk-bone metabarcoding method to investigate ancient DNA in faunal assemblages. Sci Rep 3: 3371.
- Haouchar D, Haile J, McDowell MC, Murray DC, White NE, et al. (2014) Thorough assessment of DNA preservation from fossil bone and sediments excavated from a late Pleistocenee- Holocene cave deposit on Kangaroo Island, South Australia. Quat Sci Rev 84: 56-64.
- Haile J, Froese DG, MacPhee RDE, Roberts RG, Arnold LJ et al. (2009) Ancient DNA reveals late survival of mammoth and horse in interior Alaska. Proc Natl Acad Sci USA 106: 22352-22357.
- Marrero P, Oliveira P, Nogales M (2004) Diet of the endemic Madeira Laurel Pigeon Columba trocaz in agricultural and forest areas: implications for conservation. Bird Conser Inter 14: 165-172.
- McInnis ML, Vavra M, Krueger WC (1983) A comparison of 4 methods used to determine the diets of large herbivores. J of Ran Man 36: 700-709.
- Fry B (2006) Stable Isotope Ecology. Springer-Verlag, New York City, New York.
- Soininen EM, Valentini A, Coissac E, Miquel C, Gielly L, et al. (2009) Analysing diet of small herbivores: the efficiency of DNA barcoding coupled with high-throughput pyrosequencing for deciphering the composition of complex plant mixtures. Front Zool 6: 16.
- Valentini A, Miquel C, Nawaz MA, Bellemain E, Coissac E, et al. (2009) New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Mol Ecol Resour 9: 51-60.
- Deagle BE, Chiaradia A, McInnes J, Jarman SN (2010) Pyrosequencing faecal DNA to determine diet of little penguins: is what goes in what comes out? Conserv Genet 11: 2039-2048.
- Murray DC, Bunce M, Cannell BL, Oliver R, Houston J, et al. (2011) DNA-Based Faecal Dietary Analysis: A Comparison of qPCR and High Throughput Sequencing Approaches. PLoS ONE 6(10): e25776.
- Shehzad W, Riaz T, Nawaz MA, Miquel C, Poilot C, et al. (2012) Carnivore diet analysis based on next generation sequencing: application to the leopard cat (Prionailurusbengalensis) in Pakistan. Mol Ecol 21: 1951-1965.
- Deagle BE, Jarman SN, Coissac E, Pompanon F, Taberlet P (2014) DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biol Lett 10: 0562.
- De Barba M, Miquel C, Boyer F, Mercier C, Rioux D, et al. (2013) DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet. Mol Ecol Resour 14: 306-323.
- Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics, 13: 341.
- Deagle BE, Thomas AC, Shaffer AK, Trites AW, Jarman SN (2013) Quantifying sequence proportions in a DNA-based diet study using Ion Torrent amplicon sequencing: which counts count?. Mol Ecol Resour 13: 620-633.
- Berry D, Mahfoudh KB, Wagner M, Loy A (2011) Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol 77: 7846-7849.
- Herder JE, Valentini A, Bellemain E, Dejean T, van Delft CWJJ, et al. P(2014). Environmental DNA-a review of the possible applications for the detection of invasive species. Stitching RAVON, Nijmegen. Report 2013-14.
- Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3: 294-299.
- Leray M, Yang JY, Meyer CP, Mills SC, Agudelo N, et al. (2013) A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front Zool 10: 34.
- Zeale MRK, Butlin RK, Barker GL, Lees DC, Jones G (2011) Taxon-specific PCR for DNAbarcoding arthropod prey in bat faeces. Mol Ecol Resour 11: 236-244.
- Coghlan ML, White NE, Murray DC, Houston J, Rutherford W, et al. (2013) Metabarcoding avian diets at airports: implications for birdstrike hazard management planning. Investigative Genetics 4: 27.
- Ji Y, Ashton L, Pedley SM, Edwards DP, Tang Y, et al. (2013) Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol Lett 16: 1245-1257.
- Liu S, Li Y, Lu J, Su X, Tang M, et al. (2013) SOAPBarcode:revealing arthropod biodiversity through assembly of Illumina shotgun sequences of PCR amplicons. Methods Ecol Evol 4: 1142-1150.
- Riaz T, Shehzad W, Viari A, Pompanon F, Taberlet P, et al. (2011) ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res 39: e145.
- Kelly RP, Port JA, Yamahara KM, Crowder LB (2014) Using Environmental DNA to Census Marine Fishes in a Large Mesocosm. PLoS ONE 9: e86175.
- Cooper A: DNA from museum specimens. New York: Springer; 1994.
- Cooper A, Lalueza-Fox C, Anderson S, Rambaut A, Austin J, et al. (2001) Complete mitochondrial genome sequences of two extinct moas clarify ratite evolution. Nature 409: 704-707.
- Murray DC, Haile J, Dortch J, White NE, Haouchar D, et al. (2013) Scrapheap challenge: a novel bulk-bone metabarcoding method to investigate ancient DNA in faunal assemblages. Sci Rep 3: 1-8.
- Taylor PG (1996) Reproducibility of ancient DNA sequences from extinct Pleistocene fauna. Mol Biol Evol 13: 283-285.
- Deagle BE, Gales NJ, Evans K, Jarman SN, Robinson S, et al. (2007) Studying Seabird Diet through Genetic Analysis of Faeces: A Case Study on Macaroni Penguins (Eudyptes chrysolophus). PLoS ONE 2(9): e831.
- Herlemann DPR, Labrenz M, Juergens K, Bertilsson S, Waniek JJ, et al. (2011) Transition in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5: 1571-1579.
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, et al. 2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1.
- Taberlet P, Coissac E, Pompanon F et al. (2007) Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 35: e14.
- Quéméré E, Hibert F, Miquel C, Lhuillier E, Rasolondraibe E, et al. (2013) A DNA Metabarcoding Study of a Primate Dietary Diversity and Plasticity across Its Entire Fragmented Range. PLoS ONE 8(3): e58971.
- Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mor Bio 17: 1105-1109.
- Chen S, Yao H, Han J, Liu C, Song J, et al. (2010) Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One. 5:e8613.
- Richardson RT, Lin CH, Sponsler DB, Quijia JO, Goodell K, et al. (2015) Application Of Its2 Metabarcoding To Determine The Provenance Of Pollen Collected By Honey Bees In An Agroecosystem. Appl Plant Sci 3(1). pii: apps.1400066.
- Baamrane MAA, Shehzad W, Ouhammou A, Abbad A, Naimi M, et al. (2012) Assessment of the food habits of the Moroccan dorcas gazelle in M’Sabih Talaa, West central Morocco, using the trnL approach. PLoS ONE 7: e35643.
- White TJ, Bruns T, Lee S, Taylor JW (1990)Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR protocols: a guide to methods and applications. Edited by Innis MA, Gelfand DH, Sninsky JJ, White TJ, Innis MA, Gelfand DH, Sninsky JJ, White TJ. New York, N.Y: Academic Press, Inc; 315-322.
- Geml J, Gravendeel B, van der Gaag KJ, Neilen M, Lammers Y, et al. (2014) The Contribution of DNA Metabarcoding to Fungal Conservation: Diversity Assessment, Habitat Partitioning and Mapping Red-Listed Fungi in Protected Coastal Salix repens Communities in the Netherlands. PLoS ONE 9(6): e99852.
- Hardy CM, Krull ES, Hartley DM, Oliver R (2010) Carbon source accounting for fish using combined DNA and stable isotope analyses in a regulated lowland river weir pool. Mol Ecol 19: 197-212.
- Blaxter ML, De Ley P, Garey JR , Liu LX, Scheldeman P, et al. (1998) A molecular evolutionary framework for the phylum Nematoda. Nature 392: 71-75.
- Creer S, Fonseca VG, Porazinska DL, Giblin-Davis RM, Sung W, et al. (2010) Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Mol Ecol 19: 4-20.