Case Report
Paraneoplastic Adenocarcinoma Presenting as Bulbar-Predominant Motor Neuronopathy: A Reversible ALS Mimic in an Elderly Male
Fatema Arsiwala1, Akansha Joshi1*, Ashutosh Tiwari1, Aarohi Doshi2, Tushar Raut2 and Jyotsna Oak3
1Department of General Medicine, Kokilaben Dhirubhai Ambani Hospital, Andheri, Mumbai, Maharashtra, India
2Department of Neurology, Kokilaben Dhirubhai Ambani Hospital, Andheri, Mumbai Maharashtra, India
3Head of Department, General Medicine and Consultant Rheumatologist, Kokilaben Dhirubhai Ambani Hospital, Andheri, Mumbai, Maharashtra, India
2Department of Neurology, Kokilaben Dhirubhai Ambani Hospital, Andheri, Mumbai Maharashtra, India
3Head of Department, General Medicine and Consultant Rheumatologist, Kokilaben Dhirubhai Ambani Hospital, Andheri, Mumbai, Maharashtra, India
*Corresponding author:Akansha Joshi, Department of General Medicine, Kokilaben Dhirubhai Ambani Hospital Andheri, Mumbai, Maharashtra, India. E-mail Id: akankshajoshi710@gmail.com
Article Information:Submission: 07/02/2026; Accepted: 03/03/2026; Published: 05/03/2026
Copyright: © 2026 Arsiwala F, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Background:Bulbar-onset amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder, characteristically non-reversible. In rare instances, paraneoplastic neurological syndromes (PNS) can present with ALS-like features. These immune-mediated conditions may show modest response to
immunotherapy but typically require tumor therapy for meaningful recovery.
Case Presentation:A 75-year-old man presented with two months of progressive dysphagia, muscular twitching of the tongue, and weakness. Neurophysiology confirmed motor neuronopathy. PET CT revealed a carotid bifurcation mass and hypermetabolic rectosigmoid thickening. Biopsy of a cervical lymph node demonstrated moderately differentiated adenocarcinoma. Intravenous corticosteroids resulted in only partial improvement, but subsequent targeted radiotherapy to the primary malignancy led to marked and sustained recovery of bulbar and motor function.
Conclusion:This case highlights the diagnostic importance of identifying paraneoplastic motor neuronopathies in atypical, rapidly progressive bulbar presentations. ALS mimics may respond incompletely to corticosteroids but show more substantial benefit with targeted oncologic treatment.
Case Presentation:A 75-year-old man presented with two months of progressive dysphagia, muscular twitching of the tongue, and weakness. Neurophysiology confirmed motor neuronopathy. PET CT revealed a carotid bifurcation mass and hypermetabolic rectosigmoid thickening. Biopsy of a cervical lymph node demonstrated moderately differentiated adenocarcinoma. Intravenous corticosteroids resulted in only partial improvement, but subsequent targeted radiotherapy to the primary malignancy led to marked and sustained recovery of bulbar and motor function.
Conclusion:This case highlights the diagnostic importance of identifying paraneoplastic motor neuronopathies in atypical, rapidly progressive bulbar presentations. ALS mimics may respond incompletely to corticosteroids but show more substantial benefit with targeted oncologic treatment.
Keywords:Amyotrophic Lateral Sclerosis; Paraneoplastic Neurological Syndrome; Motor Neuronopathy; Adenocarcinoma; Bulbar Dysfunction; Radiotherapy
Introduction
Motor neuron diseases, especially ALS, are progressive
neurodegenerative conditions without disease-modifying therapy.
Bulbar-onset ALS is characterized by early dysphagia and cranial
nerve involvement and typically follows an inexorably downhill
course. Crucially, ALS is not responsive to corticosteroids or
immunotherapy.
Paraneoplastic neurological syndromes (PNS), though rare, may phenocopy ALS by producing motor neuronopathy in the context of malignancy. These conditions are immunologically mediated and may stabilize or improve following immunomodulation or tumordirected therapy. We describe a case of adenocarcinoma-associated motor neuronopathy with bulbar predominance that showed only partial steroid responsiveness but much stronger sustained improvement with radiotherapy.
Paraneoplastic neurological syndromes (PNS), though rare, may phenocopy ALS by producing motor neuronopathy in the context of malignancy. These conditions are immunologically mediated and may stabilize or improve following immunomodulation or tumordirected therapy. We describe a case of adenocarcinoma-associated motor neuronopathy with bulbar predominance that showed only partial steroid responsiveness but much stronger sustained improvement with radiotherapy.
Case Presentation
History:
A 75-year-old retired male presented to us with a two-month
history of progressive dysphagia (initially to solids, later to liquids),
frequent choking, excessive salivation, and weak cough. Over the
same period, he developed generalized weakness and imbalance of
gait, requiring support.
Two weeks prior to onset of dysphagia, he had a painful right
cervical swelling and a dry cough. This was treated with antibiotics
providing only temporary relief.
Past medical history: He was a known hypertensive for 10 years,
and had recently been diagnosed with type 2 diabetes mellitus. He
had no prior malignancy or neurological disorder.
His examination revealed the following:
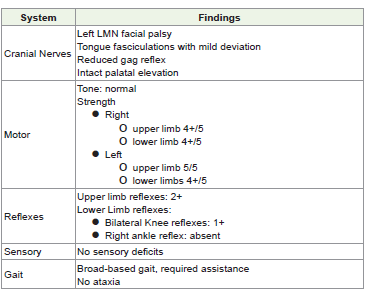
• Cranial nerves: Left LMN facial palsy, fibrillations in the
tongue with mild deviation, reduced gag reflex, intact palatal
elevation, muscular fasciculations in the upper back
• Motor: Tone normal; strength – right upper limb 4+/5, left
upper limb 5/5, both lower limbs 4+/5.
• Reflexes: Upper limb reflexes 2+, knees 1+, right ankle absent.
• Gait: Broad-based, required assistance. No ataxia or sensory
deficits
Investigation findings are outlined in [Table 1].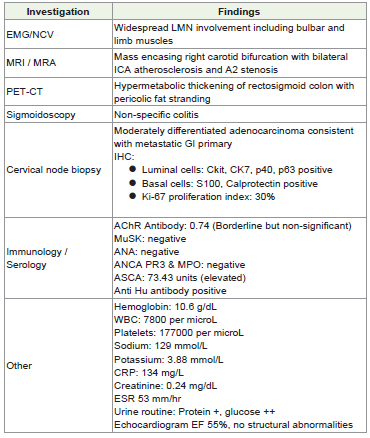
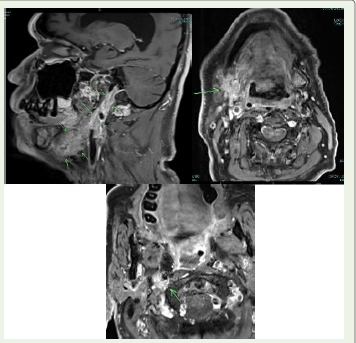
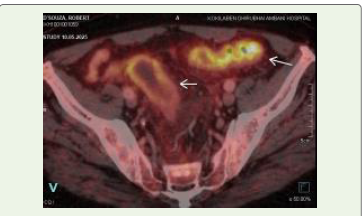
Clinical course:
Given high aspiration risk, an NG tube was inserted. He was
administered intravenous methylprednisolone (1 g/day × 5 days).
This led to modest improvement in swallowing and energy, but
deficits persisted.Following confirmation of adenocarcinoma, the patient underwent targeted radiotherapy to the suspected rectosigmoid primary and regional nodes. In the weeks after radiotherapy, his bulbar function, swallowing, and gait improved significantly, and he was able to resume partial oral feeding. This dual pattern suggested an immune-mediated, paraneoplastic motor neuronopathy, partially steroid-responsive but substantially improved only after oncologic treatment.
The patient was followed up for 12 months after completion of radiotherapy. At 3 months post treatment, he demonstrated marked improvement in bulbar function, with significant reduction in dysphagia and aspiration episodes, allowing removal of the nasogastric tube and gradual resumption of oral feeding. Tongue fasciculations diminished, facial weakness improved, and limb strength improved to near-baseline (Medical Research Council grade 5-/5 proximally and distally). By 6 months, he was independently ambulatory without support and had regained functional swallowing with only mild residual fatigue on prolonged speech. At the 12-month follow-up, neurological status remained stable without progression of motor neuron signs. There was no emergence of upper motor neuron features. This sustained neurological stabilization and functional recovery over one year strongly supports a paraneoplastic etiology rather than classical ALS, which would be expected to show relentless progression.
Discussion
Bulbar-onset amyotrophic lateral sclerosis (ALS) is classically
characterized by relentless progression, mixed upper and lower
motor neuron involvement, and absence of meaningful response to
immunotherapy or oncologic treatment. In contrast, paraneoplastic
neurological syndromes (PNS) are immune-mediated disorders
that may mimic ALS but differ fundamentally in pathophysiology,
prognosis, and therapeutic responsiveness.
Several features in this case strongly favored a paraneoplastic motor neuronopathy over classical ALS. The temporal profile was subacute, progressing over weeks rather than months to years. Neurological findings were predominantly lower motor neuron in distribution, without upper motor neuron signs, and remained nonprogressive over 12 months of follow-up. Anti-Hu antibody positivity provided serological evidence of a high-risk paraneoplastic antibody associated with malignancy-related neurological syndromes. There was a clear temporal association between neurological onset and the diagnosis of metastatic adenocarcinoma. Most importantly, the patient demonstrated partial responsiveness to corticosteroids and marked, sustained improvement following definitive tumor directed radiotherapy, an outcome incompatible with the natural history of classical ALS.
immunotherapy providing only transient stabilization in paraneoplastic motor neuron disease, [1] partial improvement or stabilization in a subset of patients with immune modulation,[2] resolution of motor neuronopathy following resection of colon adenocarcinoma,[3] and reversible ALS-like presentations where recovery followed treatment of the underlying malignancy.[5,6] Thus, our case confirms that tumor-directed therapy, such as surgery, chemotherapy, or radiotherapy, are often decisive in achieving sustained neurological recovery.
Several features in this case strongly favored a paraneoplastic motor neuronopathy over classical ALS. The temporal profile was subacute, progressing over weeks rather than months to years. Neurological findings were predominantly lower motor neuron in distribution, without upper motor neuron signs, and remained nonprogressive over 12 months of follow-up. Anti-Hu antibody positivity provided serological evidence of a high-risk paraneoplastic antibody associated with malignancy-related neurological syndromes. There was a clear temporal association between neurological onset and the diagnosis of metastatic adenocarcinoma. Most importantly, the patient demonstrated partial responsiveness to corticosteroids and marked, sustained improvement following definitive tumor directed radiotherapy, an outcome incompatible with the natural history of classical ALS.
immunotherapy providing only transient stabilization in paraneoplastic motor neuron disease, [1] partial improvement or stabilization in a subset of patients with immune modulation,[2] resolution of motor neuronopathy following resection of colon adenocarcinoma,[3] and reversible ALS-like presentations where recovery followed treatment of the underlying malignancy.[5,6] Thus, our case confirms that tumor-directed therapy, such as surgery, chemotherapy, or radiotherapy, are often decisive in achieving sustained neurological recovery.
Alternative explanations:
Although our patient demonstrated widespread LMN
involvement consistent with a paraneoplastic motor neuronopathy,
two features, that is the presence of a carotid bifurcation lesion and
an LMN facial palsy, could suggest direct tumor infiltration of cranial
nerves. Local compressive or infiltrative effects remain an important
differential diagnosis in patients with focal adenopathy.Paraneoplastic etiology remains more likely in our case rather than an isolated local invasion due to the following several points:
1. The distribution of LMN signs extended beyond the cranial territory of the cervical lesion, involving limb muscles and bulbar regions simultaneously.
2. The modest but clear response to corticosteroids suggested an immune-mediated component, which would not be expected with purely infiltrative pathology.
3. Most importantly, the marked and sustained functional recovery after targeted radiotherapy to the primary malignancy and nodal disease is characteristic of paraneoplastic neurological syndromes, in which definitive oncological therapy often results in improvement.
A limitation of this report is the absence of a comprehensive paraneoplastic antibody panel (Hu, CV2, KLHL11, amphiphysin, etc.), which might have provided further serological support. Nonetheless, the clinical trajectory—rapid progression, partial steroid responsiveness, and dramatic oncological treatment response— supports a paraneoplastic etiology over ALS or local infiltration alone.
Conclusion
This case highlights adenocarcinoma-associated paraneoplastic
motor neuronopathy mimicking bulbar-onset ALS. Unlike ALS, such
cases may show partial steroid responsiveness and more substantial
recovery with targeted oncological therapy. Clinicians should
suspect PNS in rapidly progressive bulbar syndromes, as timely
recognition and combined immune- and tumor-directed therapy
may dramatically alter prognosis.
Rapidly progressive bulbar ALS-like presentations in elderly patients should raise suspicion for paraneoplastic syndromes. Partial response to corticosteroids can occur, but sustained recovery usually requires oncological treatment.
Direct tumor infiltration should be considered, but widespread LMN involvement supports a systemic immune-mediated mechanism.
Timely recognition can significantly alter prognosis in ALS mimics.
Rapidly progressive bulbar ALS-like presentations in elderly patients should raise suspicion for paraneoplastic syndromes. Partial response to corticosteroids can occur, but sustained recovery usually requires oncological treatment.
Direct tumor infiltration should be considered, but widespread LMN involvement supports a systemic immune-mediated mechanism.
Timely recognition can significantly alter prognosis in ALS mimics.
Patient Consent and Ethics:
Written informed consent for publication was obtained from the
patient’s next of kin. Institutional ethics approval was waived as per
local policy for single case reports.References
Citation
Arsiwala F, Joshi A, Tiwari A, Doshi A, Raut R, et al. Paraneoplastic Adenocarcinoma Presenting as Bulbar-Predominant Motor Neuronopathy: A Reversible ALS Mimic in an Elderly Male. Indian J Neurol. 2026;7(1): 163.