Case Report
Waldenstorm’s Macroglobulinemia: The Great Masquerader: A Case Report
Chakraborty S1*, Mishra A and Sarkar S2
1Department.of Medicine, Sri Ramachandra Medical College and Hospital (SCBMCH), Cuttack, India
2Department of Radiodiagnosis, Sri Ramachandra Medical College and Hospital, Cuttack, India
2Department of Radiodiagnosis, Sri Ramachandra Medical College and Hospital, Cuttack, India
*Corresponding author:Shayri Chakraborty, Department of General Medicine, S.C.B. Medical college and Hospital, Cuttack, Odisha, India. Email id: shayri1012@gmail.com
Article Information:Submission: 18/05/2025; Accepted: 09/06/2025; Published: 10/06/2025
Copyright: © 2025 Chakraborty S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Waldenström’s Macroglobulinemia (WM) is a slow growing hematological malignancy characterized by proliferation of plasma cells, lymphocytes and plasmacytoid lymphocytes in bone marrow secreting monoclonal immunoglobulin IgM and a risk of hyper viscosity syndrome. This case report highlights a
unique presentation observed in a 60 years old farmer with neuropathy, and necrotizing soft tissue infection as the primary manifestation of WM. Notably, our patient had recurrent spontaneous hemolysis and presented to us a second time with Acrocyanosis, prompting for evaluation of cold agglutinin disease.This
led to the final diagnosis of WM. Treatment was commenced with Bendamustine, Rituximab and Dexamethasone led to complete response (normalization of serum IgM and absence of bone marrow and extramedullary disease). As of the publication of this report, patient is in remission. In our case the patient had manifestations of symmetric peripheral neuropathy which predated all other manifestations, evaluation for the same might have prevented the potentially life-threatening necrotising infection he subsequently presented with.
Keywords:Waldenstorm’s Macroglobulinemia; Cold Agglutinins; Peripheral Neuropathy; Necrotising Soft Tissue Infection; Autoimmune HaemolyticAnaemia; ImmunoglobulinM Monoclonal Gammopathy
Patient Description
Case of a previously healthy 60-year-old farmer in Odisha
presented with swelling, redness and local rise of temperature of distal
extremities of upper and lower limbs to SCB MCH, Cuttack during
the month of December. A month prior he had started developing
complaints of distal symmetric paranesthesia (numbness and pins
and needle sensation) in his upper and lower limbs, which was
unprovoked and constant for which and he had not sought medical
care.Two weeks prior seeking medical care he had a spontaneous
painless ulceration over his scrotum. At presentation the ulcer had a
clear base and margins appeared gangrenous. This was not associated
with fever, prior history of insect bite, discharge per urethra. Over the
course of a week he then developed fever and was treated at a local
hospital for which he received empirical antibiotics.The following
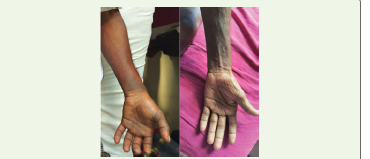
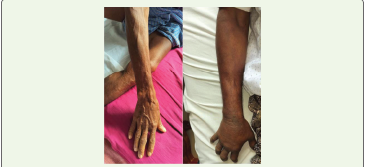
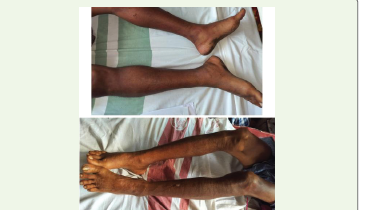
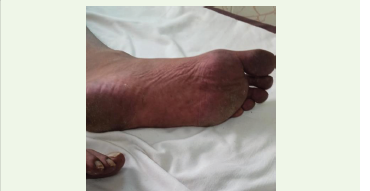
week he suddenly developed swelling and erythematous discoloration
of his hands and feet which prompted him to seek medical care in our
hospital.
Provisional Diagnosis
He had no prior medical or family history or toxin exposure.
Within a day following admission the discolouration had rapidly
spread to extend into mid- forearms and mid-calf. Prompt arterial
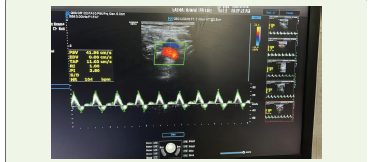
and venous doppler was done which showed patent vessels with
normal flow, however muscular plains showed soft tissue oedema
and heterogeneous density, a provisional diagnosis of necrotising
soft tissue infection was made and empirical antibiotics was
initiated with Intravenous (IV) Vancomycin, IV Meropenem and
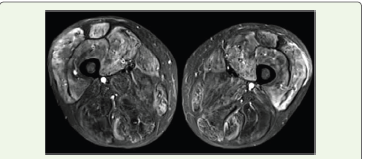
IV Clindamycin.Creatinine Phosphokinase (CPK) was 63,100 mg/dl
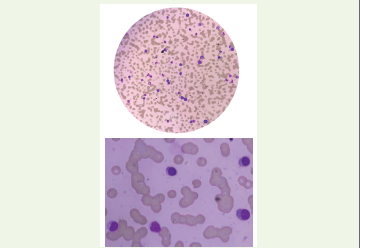
and Myonecrosis was confirmed via MRI. Bone marrow aspiration
showed lymphoplasmatic cells and plasma cells constituting 20% of
all Mononuclear cells.
Multiple Myeloma thus was ruled out due to absence of greater
than 60% clonal plasma cells and absence of myeloma defining events,
and light chains Serum involved / uninvolved<100. Bedside blood
samples collected post admission underwent recurrent spontaneous
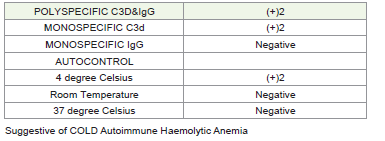
hemolysis and thus under suspicion of cryoglobulinemia, DCT- ICT
was sent: DCT positive [4+] for cold agglutinins {titre>1:1024}.
Due to the constellation of presentations of neuropathy,
ulceration and Necrotising soft tissue infection (NSTI) and cold
agglutination a provisional diagnosis of secondary Cold agglutination
disease, secondary to lymphoproliferative disorders, monoclonal
gammopathyor autoimmune disorders was considered.
Evaluation and Investigations:
Bed-side evaluation showed a discreet solitary inguinal
lymphadenopathy which was however not amenable for biopsy,
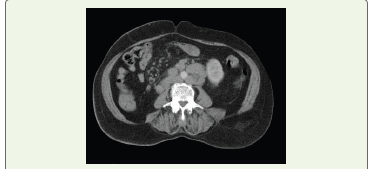
USG showed, hepato- splenomegaly and multiple enlarged peri
pancreatic and para-portal lymph nodes, which were aspirated and
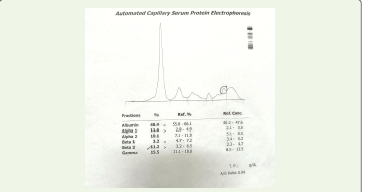
had evidence of reactive hyperplasia. Serum electrophoresis was
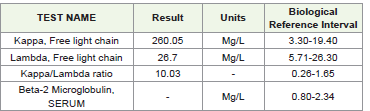
negative for M band, however increased kappa light chains 354.28mg/
L[3.30mg/L-19.40 mg/L] tested on SPAplus software. K/l ratio: 9.87
seen on immunofixation. In addition, viral marker for HCV was
negative (Table 2), Swab culture from scrotal ulceration was sterile,
peripheral blood sample drawn prior commencement of antibiotic
was sterile, Serum Procalcitonin was negative, and Bone marrow
aspiration showed erythroid hyperplasia with lymphoplasmacytic
cells consisting of 20% of mononuclear cells.Despite resolution of symptoms there was persistent motor
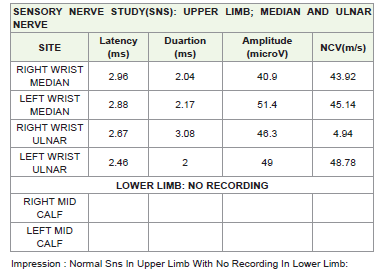
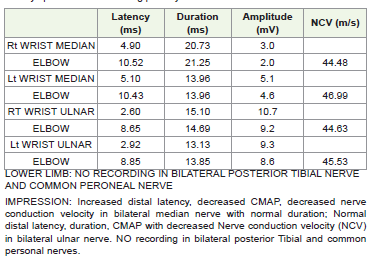
weakness. Nerve conduction study (NCS) showed evidence of
motor-sensory axonal demyelinating neuropathy of all limbs.
There was gradual response to antibiotic regimen with reduction of
inflammation and over the course of two weeks. Over the course of
a month the patient had improved with no constitutional symptoms
however had persistent motor weakness in distal extremities and was
ambulatory with support. At this stage he was discharged on request
and advised follow up at two weeks with and to review sos.
Provisional diagnosis of lymphoma vs primary CAD:
A week following discharge he had complaints of painless
persistent erythema of dorsum of bilateral feet, and hands, and had
sought tele -consultation. Features were suggestive of acrocyanosis;
refusing admission patient presented to follow up after a month.He showed progressive thrombocytopenia in serial CBC; at
this stage he presented with a fresh attack of acrocyanosis and cold
AIHA was confirmed. Repeat electrophoresis showed presence of
m-spike with increase in k/l ratio 10.03; kappa light chain 268.03mg/L
[3.30mg/L -19.40 mg/L] tested on SPA plus. Follow up of lymph node
examination showed increase in size in the inguinal node. Excision
biopsy of inguinal lymphnode was suggestive of non-Hodgkin
lymphoma.
Treatment:
Adiagnosis low grade lymphoma with secondary CAD was
done at this stage and a cycle of BR [Bedamustine-Rituximab] was
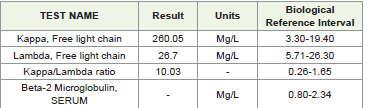
commenced. A month later on followup are peat electrophoresis
showed 214.73[3.30mg/L -19.40 mg/L] tested on SPA plus: K/L ratio
14.56 (rising trend) M-spike present with IgM component. Repeat
bone marrow aspiration showed presence of lymphoplasmacytic
cells; plasma cells >10%.Diagnosis was revised to WM and patient was commenced on
BRD regimen and after 6 cycles of therapy is currently in remission
with no symptoms of hyper viscosity subsequent to initiation of
treatment.
Follow-up and outcomes:
As of the publication of this report, patient is in remission.
His neurological defect has sufficiently recovered that he is now
ambulatory with support of a walking stick. No further complications
were encountered during treatment duration or subsequently
thereafter.Discussion
Waldenstorm’s macroglobulinemia (WM) or lymphoplasmacytic
leukemia is a rare hematological malignancy and poses a challenging
diagnosis due to lack of specific immunopathology. The diagnostic
criteria of thedisease are:
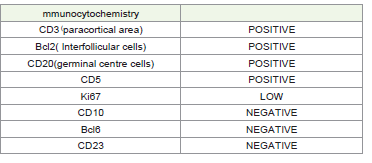
1. Presence of IgM monoclonal gammopathy of any size 2. Greaterthan10% of biopsy sample must demonstrate infiltration by small lymphocytes that exhibit plasmacytoid or plasma cell differentiation 3. Infiltrate should express typical immunophenotype: surface IgM+,CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, CD5 variable, CD10-, CD23-, CD103-, and CD108-.
1. Presence of IgM monoclonal gammopathy of any size 2. Greaterthan10% of biopsy sample must demonstrate infiltration by small lymphocytes that exhibit plasmacytoid or plasma cell differentiation 3. Infiltrate should express typical immunophenotype: surface IgM+,CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, CD5 variable, CD10-, CD23-, CD103-, and CD108-.
The presentation of WM is a constellation of hyper-viscosity [1-4]
autoimmune Hemolysis, and neuropathy.
This patient primarily presented with sensory neuropathy and
necrotising soft tissue infection, the patient had had no prior history
of hyper-viscosity or symptomatic anemia, there have been case
reports of gangrenous necrosis associated with WM [4,5] expansive
limb gangrene, bulbous cellulitis and gangrenous cholecystitis have
also been reported. [16] In our case, recurrent hemolysis led to the
suspicion of cold agglutinins, positive DAT (direct agglutination
test), specific DAT for anti- C3d [4+] reactive at 4degrees with titre
>1:1024 was diagnostic.
The occurrence of Cold agglutinin mediated hemolysisis a
recognised entity in WM, however <5% will have Cold agglutinin
disease (CAD) [7]. In WM, the monoclonal IgM para-proteins can
present with cold auto immune hemolyticanaemia (AIHA).This Cold
agglutination syndrome can occur secondary to CAD associated
B cell neoplasms, marginal zonelymphoma, small lymphocytic
lymphomaor LPL seen in WM isto be differentiated from Cold
agglutination disease. Presence of marrow lymphoplasmacytic cells
and the L265P mutation in the MYD88 gene is highly characteristic of
WM. In a series of 232patientsreviewedin202 pathologists identified
up to14% reported cases of CAD were due to WM. [23].
The IgM para proteins are directed against RBC antigens most commonly the I antigen. These then cause complement mediated extravascular haemolysis, however, during periods of stress intravascular hemolysis may be precipitated. The forces enabling antigen binding are characteristically weak, made stronger in cold temperatures due to reduced Brownian movement accounting for the cold reactivity.
The IgM para proteins are directed against RBC antigens most commonly the I antigen. These then cause complement mediated extravascular haemolysis, however, during periods of stress intravascular hemolysis may be precipitated. The forces enabling antigen binding are characteristically weak, made stronger in cold temperatures due to reduced Brownian movement accounting for the cold reactivity.
Cryoglobulins in contrast are cold precipitated immunoglobulins
which do not bind to RBC surface. In WM both spectrums of disease
are encountered where the large polyvalent IgM pentamerises and
causes vascular occlusion leading to phenomenon of acrocyanosis as
seen in our patient.
Due to the aberrant immunoglobulins along with associated hypogammaglobulinemia and reduced IgA and IgG infections, are commonly seen in WM, notably bacterial and respiratory tract infections. [17-18]In the past invasive fungal infections have also been reported. In this spectrum of paraproteinemias MGUS and MM increased risk of bacterial and viral infections [15]; there are reported cases of increased incidence of necrotisings of tissue infections [17,19] Most common findings in cases of NSTI as per reports are those of swelling (75%),erythema and pain beyond site of erythema; all of which were reported in our case on presentation.[20]
Early suspicion and prompt antibiotic initiation is crucial to reducesubsequent mortality and morbidity. We established Myonecrosis due the presence of heterogenous muscle density on USG limb which was then followed by MRI of the affected parts, an elevated CPK enzyme and resolution of infection as per empirical antibiotic initiation on meropenem, vancomycin and clindamycin. The LRINEC score is commonly used to differentiate NSTI from other soft-tissue infection scan help clinical decision between a conservative management (likeourcase) vs a surgical one. However, this core poses a challenge in neutropenic patients as in those that might been countered in MM/WM. While radiological evidence can suggest NSTI a strong clinical suspicion is required for early diagnosis in the disease course [20-22]. Notably, NSTI has also been reported with the treatment of Lenalidomidein MM in the past [19].
Due to the aberrant immunoglobulins along with associated hypogammaglobulinemia and reduced IgA and IgG infections, are commonly seen in WM, notably bacterial and respiratory tract infections. [17-18]In the past invasive fungal infections have also been reported. In this spectrum of paraproteinemias MGUS and MM increased risk of bacterial and viral infections [15]; there are reported cases of increased incidence of necrotisings of tissue infections [17,19] Most common findings in cases of NSTI as per reports are those of swelling (75%),erythema and pain beyond site of erythema; all of which were reported in our case on presentation.[20]
Early suspicion and prompt antibiotic initiation is crucial to reducesubsequent mortality and morbidity. We established Myonecrosis due the presence of heterogenous muscle density on USG limb which was then followed by MRI of the affected parts, an elevated CPK enzyme and resolution of infection as per empirical antibiotic initiation on meropenem, vancomycin and clindamycin. The LRINEC score is commonly used to differentiate NSTI from other soft-tissue infection scan help clinical decision between a conservative management (likeourcase) vs a surgical one. However, this core poses a challenge in neutropenic patients as in those that might been countered in MM/WM. While radiological evidence can suggest NSTI a strong clinical suspicion is required for early diagnosis in the disease course [20-22]. Notably, NSTI has also been reported with the treatment of Lenalidomidein MM in the past [19].
Nerve conduction studies conducted showed distal symmetric
axonal- demyelination neuropathy in all limbs. Neurological
complications are frequently found (20%) [11] in WM and
occur as a result of hyper-viscosity, immunoglobulin deposition,
direct infiltration by neoplastic lymphoplasmacytoid cells,or
transformed high-gradelymphoma cells. They most commonly
manifest as a distal, symmetric, slowly progressive sensorimotor
peripheral neuropathy causing parasthesias and weakness
[12]. The neuropathy in WM is usually demyelinating but
certain studieshavereportedanaxonalpredominantpattern [13].
ParaproteinsIgM can act as autoantibody directed against myelin
associated glycoproteins or other nerve components resulting
in neuropathy [5,6] this resultsinadistalsymmetricprogressive
demyelination sensory predominant neuropathy; motor axonal
neuropathy Is more Common in disease like POEMS and
amyloidosis. Other Neurological manifestations include cranial
nerve palsies, mononeuropathy, mononeuritis multiplex, multifold
leukoencephalopathy or infiltration of CNS (Bing-Neel syndrome)
[14]. These symptoms show improvement with therapy but it has
been reported that some degree of residual neuropathy still persists.
Treatment:
Asymptomatic cases of WM donot warrant treatment. Treatment
is initiated in the presence of certain clinical indications like hyperviscosity,
symptomatic lymphadenopathy (size >5cm), organomegaly,
recurrent fever, night sweats, weight loss, peripheral neuropathy or
lab indications likesymptomatic cryoglobulinemia, nephropathy,
presence of hemolytic anemia, thrombocytopenia nephropathy. [8]For newly diagnosed symptomatic WM, the first line of therapy is a combination of Rituximab (375mg/m2 on D1 of each cycle) and Bendamustine (90mg/m2 on D1 and 2 of each cycle) while continuous therapy with a BTK inhibitor like Ibrutinib, Zanubrutinib (more effective with a good safety profile) is proffered for older frail patients or non-consenting individuals. [9-10].
Conclusion
WM should be considered in elderly with unexplained weakness,
neurological deficit, coagulopathy, visual difficulty and autoimmune
haemolytic anemia. Although slow growing, with expansion of
clonal LPL cells there is subsequent cytopenia manifesting as anemia,
lymphadenopathyandhepatosplenomegaly.
In our case report our patient had manifestations of symmetric peripheral neuropathy which predated all other manifestations, evaluation for the same might have prevented the potentially life threatening necrotising infection he subsequently presented with. This case report highlights the importance of considering atypical differentials when confronted with an absence of obvious alternatives.
Consent: The patient has signed a written informed consent form, agreeing to the publication of this case report and related medical images and lab reports.
In our case report our patient had manifestations of symmetric peripheral neuropathy which predated all other manifestations, evaluation for the same might have prevented the potentially life threatening necrotising infection he subsequently presented with. This case report highlights the importance of considering atypical differentials when confronted with an absence of obvious alternatives.
Consent: The patient has signed a written informed consent form, agreeing to the publication of this case report and related medical images and lab reports.
References
Citation
Chakraborty S, Mishra A, Sarkar S. Waldenstorm’s Macroglobulinemia: The Great Masquerader: A case Report. Indian J Neurol. 2025;6(1): 145.