Case Report
Mitochondrial Myopathy with Chronic Progressive External Ophthalmoplegia: A Case Report
Ishreen Ahuja1, Harshpreet Singh1 and Ranganath Honnal1
Department of General Medicine, Government, Medical College and Hospital, Chandigarh, India
*Corresponding author: Ranganath Honnal,
Department of General Medicine, Government, Medical College and Hospital,
Chandigarh , India E-mail id : honnalranganath@gmail.com
Submission: 03/04/2023; Accepted: 05/05/2023; Published: 08/05/2023
Copyright: © 2023 Ahuja I, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
A 44-year-old male patient presented with progressive ptosis. Mitochondrial Myopathy with Chronic Progressive External Ophthalmoplegia was diagnosed. Case findings along with a discussion of existing literature are presented.
Introduction
Chronic Progressive External Ophthalmoplegia (CPEO) is a
slowly progressive disorder affecting the extraocular muscles, which
was first described by Von Graefe in 1868. Initially, it was believed to
be caused by neuronal degeneration, but later studies have confirmed
that it has a myopathic origin. CPEO usually presents with ptosis in
childhood or adolescence, followed by ophthalmoparesis, and it does
not involve the ciliary and iris muscles. Both males and females are
equally affected, and the pattern of inheritance is usually autosomal
dominant, although rare recessive or uncertain cases also exist. Some
nuclear genes mutation such as POLG1, Twinkle, and ANT1 have
been implicated in CPEO. Although some cases of CPEO transmitted
in a Mendelian manner are not of mitochondrial origin. Imaging
studies usually show thin and symmetrical extraocular muscles.
Muscle biopsy remains the definitive test for diagnosis, although
Polymerase Chain Reaction (PCR) can also be used to confirm the
diagnosis [1].
Case Presentation
A gentleman aged 44 years presented with progressive bilateral
symmetrical ptosis since adolescence Figure 1. The disease was
progressive for 15 years and static for the last 5 years. He did not complain of fluctuating symptoms, diplopia, blurring of vision, gait disturbance, seizures or any other muscle group weakness. The
patient had never suffered any head/facial trauma and no family
member had any similar complaints
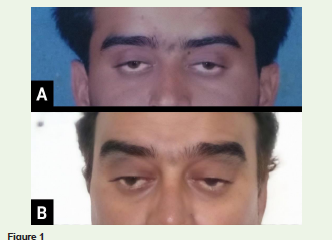
Examination revealed bilateral incomplete ptosis. Pupils were round, equal and reactive to light. Extraocular movements and facial symmetry were preserved. Fundus examination and axial and skeletal
neuromuscular examination was unremarkable.
A plain CT scan of the brain was normal. The patient had
undergone an MRI brain and bilateral orbits which also did not show
any abnormality. Thyroid Function Tests were within normal limits
and Anti-Acetylcholine receptor (AChR) antibodies were negative.
Nerve conduction studies (NCS) were normal and repeated nerve
stimulation (RNS) did not show any decremental response. Serum
creatine kinase levels were elevated (682 IU/L). Right deltoid muscle
biopsy revealed cytochrome oxidase (COX) deficient fibres and
ragged blue fibres on succinate dehydrogenase (SDH) stain. The
patient was advised to wear spectacles with eyelid supports.
Discussion
Neuromuscular causes of ptosis were ruled out with the absence of
fluctuation of symptoms, negative Anti-AChR antibodies and normal
response on RNS. Local causes were ruled on with an MRI brain
and orbit. No clinical evidence of the presence of other syndromes
like Tolosa-Hunt, Kearns-Sayre or congenital muscular dystrophies
(oculopharyngeal or myotonic dystrophy) was found.
Causes of non-progressive ophthalmoparesis are agenesis of
extraocular muscles, congenital fibrosis syndrome, and congenital
myopathies (centronuclear myopathy, central core myopathy, and
multicore myopathy) [1].
The patient presented with ptosis without extraocular muscle
weakness, which is an uncommon finding. Most patients present with
some amount of gaze abnormalities[2].
Investigations in CPEO reveal elevated creatine kinase levels and
minimal extraocular muscle volume loss despite marked weakness
[3]. Muscle biopsy shows COX deficient fibres, ragged red fibres
on Gomori Trichrome stain and ragged blue fibres on SDH and
NADH-TR stain [4]. Electron microscopy of mitochondria shows
multiple abnormal mitochondria in the subsarcolemmal region, with
paracrystalline inclusion bodies [5]. Electron microscopy and genetic
testing could not be performed on our patient.
Mitochondrial myopathies may present as isolated CPEO, CPEOplus
(CPEO with hearing loss, neuropathy, ataxia, parkinsonism, or
depression), or Kearns-Sayre syndrome. These presentations may
reflect a clinical spectrum rather than discrete illnesses [6].
Treatment includes symptomatic management with eyelid
crutches or taping. Anecdotal success without proven benefit has
been seen with coenzyme Q10, riboflavin supplementation and
ketogenic diet. Candidates for surgery are patients with preserved
bell’s phenomenon. However, surgery still carries a high risk of
exposure keratopathy even in carefully selected patients [4].
Conclusion
Mitochondrial myopathy and CPEO may be suspected in patients
who present with ophthalmoparesis with or without other muscle
group weaknesses. Progressive external ophthalmoplegia is a striking
but nonspecific clinical sign that occurs in a variety of disease states
such as myasthenia gravis, thyrotoxicosis, Guillain-Barre syndrome
and Refsum’s disease. Genetic testing has a large role in establishing
diagnosis, prognostication and genetic counselling. Although current
treatment options for CPEO are limited, continued investigation is
promising.
References
1. Schrier SA, Falk MJ (2011) Mitochondrial disorders and the eye. Curr Opin
Ophthalmol 22: 325-331.
Citation
Ahuja I, Singh H, HonnalR. Mitochondrial Myopathy with Chronic Progressive External Ophthalmoplegia: A Case Report. Indian J Neurol. 02 2023;4(1): 114.