Case Report
A Unique Case of Unilateral Treacher-Collins Syndrome with Middle Ear Aplasia: A Case Report
Anagha J, Chinmayee C*, Nikhil M, and Mahak B
Department of Radiology, LTMMC and LTMGH, Sion, Mumbai, India
*Corresponding author:Chinmayee Chitnis, Fellow, Department of Radiology, LTMMC and LTMGH, Sion, Mumbai, India, E-mail: chinmayeechitnis5694@gmail.com
Copyright: ©2024 Anagha J, et al. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Article Information:Submission: 11/12/2023; Accepted: 17/01/2024; Published: 22/01/2024
Abstract
Treacher-Collins syndrome or Mandibulofacial dysostosis (MFD) is a rare congenital disorder with autosomal dominant inheritance, associated with aberrations of craniofacial development. The most common clinical features include malar bone and mandibular hypoplasia, antimongoloid slanting of palpebral fissures, and other ear abnormalities. Computed tomography is the imaging modality of choice for the morphological analysis of the craniofacial bones in individuals with complex facial deformities and to assist in the planning of surgical intervention. We present a case of unilateral mandibulofacial dysostosis with absent middle ear ossicles.
Keywords:Treacher-Collins; Middle Ear Ossicles; Computed Tomography; Zygomatic bone
Introduction
Treacher-Collins syndrome or Mandibulofacial dysostosis (MFD)
or Franceschetti-Zwahlen-Klein syndrome is a rare congenital disorder
of craniofacial development with autosomal dominant inheritance.
Approximately 60%of cases of Treacher-Collins syndrome show a
genetic abnormality in the form of a de novo mutation involving the
treacle gene (TCOF1) on chromosome 5 with resultant interference
in the development of first and second branchial arches. Clinical
features most encountered are hypoplastic malar bone and lower
jaw, auricular abnormalities, and antimongoloid slanting of palpebral
fissures. [1] Computed tomography aids in diagnosing anatomical
abnormalities, developing surgical treatment strategy as well as postoperative
monitoring. [2] We report an interesting case of Treacher-
Collins syndrome, discussing its clinical and computed tomography
imaging features.
Case Report
A 10-month-old male, born out of a 2nd degree consanguineous
marriage and uneventful antenatal and immediate post-natal history
was brought to medical attention with complaints of deformed
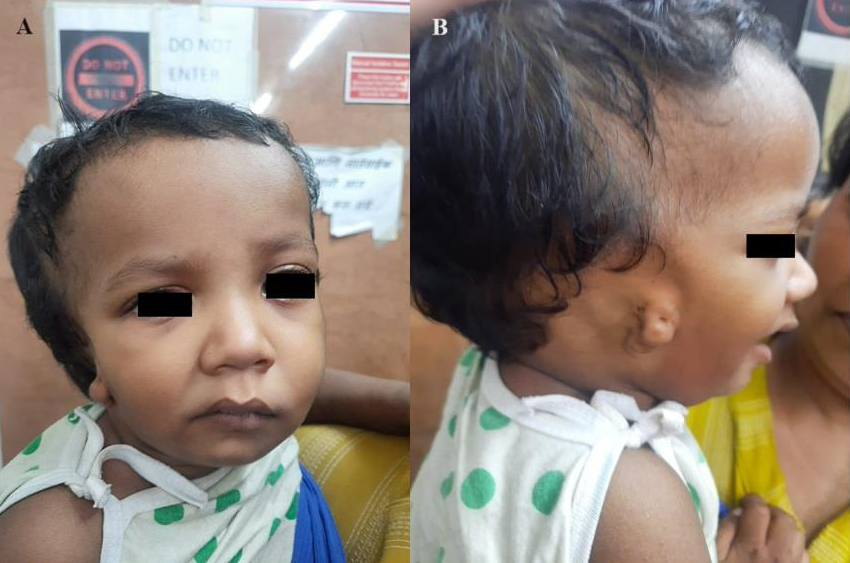
right pinna and regurgitation during feeding. On detailed clinical
examination, the child had antimongoloid slant of eyes, narrow face
with right mandibular hypoplasia, deviation of angle of mouth to the
right [Figure 1A], high arched palate, bifid uvula and retrognathism.
External ear malformation was noted in the form of grade III microtia
[Figure 1B] on the right andabsence of an external opening of the
right ear.Above features were suggestive of multiple craniofacial
abnormalities.
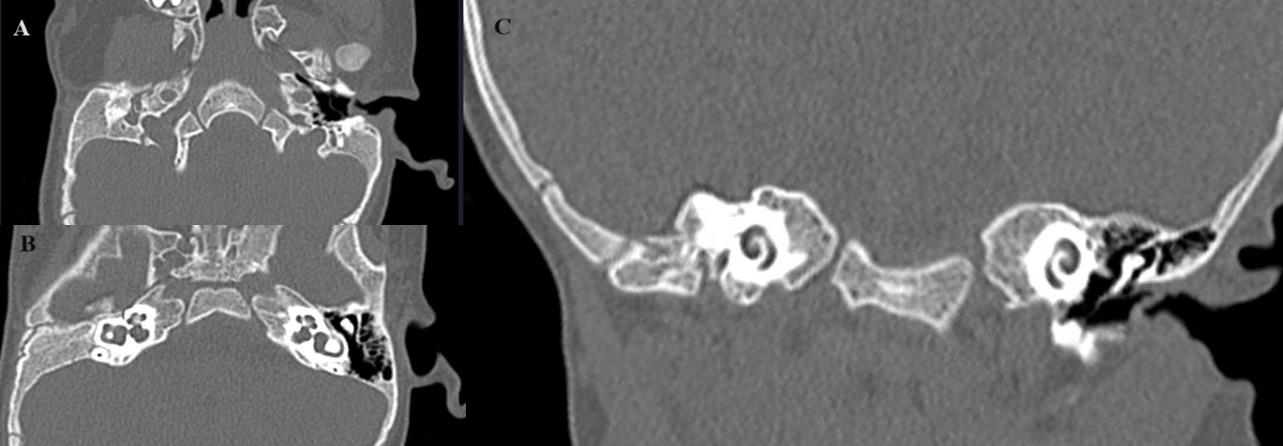
High-resolution computed tomography (HRCT)of Temporal
boneon 160 slice Multidetector CT scanner (MDCT) with thin
sections revealed,type 3 microtia and an aplastic external auditory
canal on right [Figure 2A] and [Figure 2C]. Absent right middle
ear ossicles with short and wide lateral semi-circularcanal [Figure 2B] and [Figure 2C]. The facial canal at the level of the labyrinthine
segment of facial nerve appeared narrowed as compared to left. Rest
of the facial nerve canal was not well appreciated. There was nonpneumatization
of right mastoid air cells [Figure 2A] and [Figure 2C].
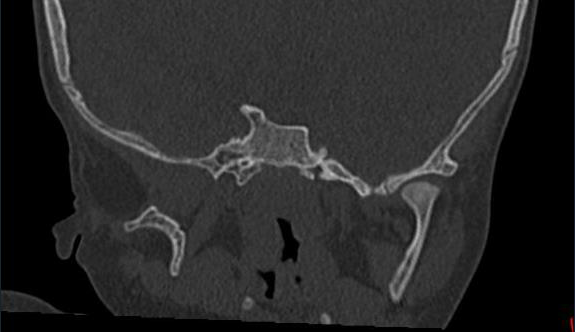
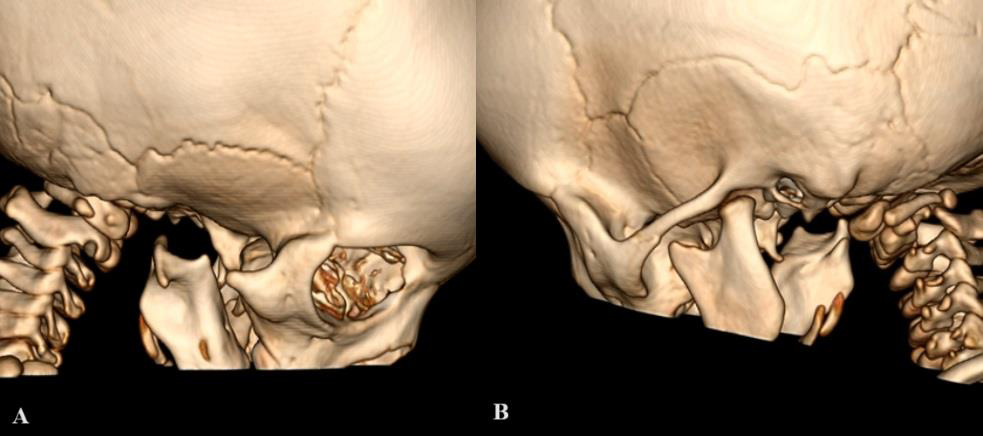
Absent right zygomatic bone and condylar process of right
mandible [Figure 4A].
Informed consent was obtained from the father of the patient
involved in the study and was informed about the potential
publication in scientific journal.
Discussion
Treacher-Collin syndrome is a disorder of craniofacial
development with an estimated incidence of 1 in 50,000 live births
with positive family history in about 40% cases and new mutations
in 60% cases.[3] A British ophthalmologist Treacher Collins first
described the features of this anomaly in 1900, however the first case
was reported nearly 54 years prior in 1846 by Thompson.The condition
was named mandibulofacial dysostosis in 1944 by Franceschetti, who
wrote a significant revision of the anomaly [1].
TCOF1 gene, which codes for serine and alanine-rich nucleolar
phosphoprotein necessary for craniofacial development is identified
as the gene responsible for this condition [4].
The clinical features originally described by Franceschetti and
Klein [5] include antimongoloid palpebral fissure, facial bone
hypoplasia (predominantly malar bone and mandible), external ear
deformity, occasional middle and inner ear abnormalities, dental
abnormalities, high arches palate, and other skeletal deformities. Our
caseshowed right sided microtia with absent external ear opening,
antimongoloid slant of eyes, hypoplasia of the right mandible with
a narrow face, right side deviated angle of mouth, high arched palate
retrognathism, and bifid uvula. They also described five clinical forms
of this syndrome: complete form, incomplete form, abortive form,
unilateral form, and atypical form. Our patient had unilateral
form with involvement only on the right side. Teberet al. defined
hypoplasia of zygomatic arch and downward slanting palpebral
fissures as minimum diagnostic criteria, both of which were seen in
our case [3].
Absent, malformed, or malposed external ears and variable
degrees of hypoplasia of the external auditory canals and middle ear
ossicles were CT findings described by Posniak et al.[6] According to
Stovin et al, the absence of a zygomatic arch was the chief radiologic
finding. [7] On the basis of CT volumetric studies, Terner et al
demonstrated that the mandibular condyle is the most common
hypoplastic structure as compared to the rest of the mandible.[8]
Loydet al in 1979 described underdeveloped and under-pneumatized
mastoid as the most obvious and constant feature in this syndrome.
[9] MDCT of our patient revealed external ear malformation in the
form of right-sided microtia and aplastic external auditory canal. All
three right middle ear ossicles were absent. The left semi-circular
canal was short and wide. Condylar process of the right mandibular
and right zygomatic bone was absent. Also, the right mastoid air cells
were non-pneumatised in our case. In our case, however, unilateral
affection was noted, only on the right side.
Facial features similar to this condition are also seen in Nager’s
syndrome, Miller’s syndrome, and Goldenhar syndrome hence must
be included as a differential diagnosis of TCS. However, preaxial limb
abnormalities and more severe mandibular hypoplasia favour Nager’s
syndrome. Miller’s syndrome can be differentiated on the basis of
ectropion and postaxial limb defectswhile features like anomalies of
vertebrae, epibulbardermoids, and asymmetrical facial anomalies are
characteristic of Goldenhar syndrome. [10,11]
For the management of this condition, a multidisciplinary
approach is necessary which involves orthodontists, craniofacial
surgeons, otolaryngologists, ophthalmologists, and speech therapists
[12].
Mutation of the gene can be identified by genetic testing, however
is expensive and hence is employed only if clinical findings are
equivocal [13].
To reduce the incidence of this syndrome, prenatal diagnosis and
genetic counseling of parents is recommended [14].
This case is unique, as it has unilateral involvement,
whereas the majority of the cases published in the literature showed
bilateral involvement. Additionally, another uncommon finding in
our case was the absence of all three middle ear ossicles.
References
Citation
Anagha J, Chinmayee C, Nikhil M, Mahak B. A Unique Case of Unilateral Treacher-Collins Syndrome with Middle Ear Aplasia: A Case Report. Indian J Appl Radiol. 2024;10(1): 189.