Review Article
Wilson’s Disease: A Brief Review withNeuroimaging Features
Paramdeep Singh 1Amarpreet Kaur 2 and Rupinderjeet Kaur3*
Corresponding author: Dr. Rupinderjeet Kaur, Assistant Professor, Department of Medicine, Guru Gobind Singh Medical College and Hospital, Baba Farid University of Health Sciences, Faridkot (Punjab), India, Ph. +91-9814030677,; E-mail: dr.rupinderjeetkaur@gmail.com
Citation: Kaur A, Kaur R. Wilson’s Disease: A Brief Review with Neuroimaging Features. Indian J Appl Radiol. 2015;1(1): 102.
Copyright © 2015 Paramdeep Singh et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Indian Journal of Applied Radiology | Volume: 1, Issue: 1
Submission: 05/05/2015; Accepted: 04/07/2015; Published: 10/07/2015
Abstract
Wilson disease is a metabolic disease resulting from defective copper metabolism leading to deposition of copper in liver, brain and eyes. Gene involved is ATP7B on chromosome 13. Systemic involvement presents in form of hepatitis, fulminant hepatic failure, cirrhosis, dysarthria, dystonia, spasticity, seizures, behavioral changes, psychosis, hemolytic anemia, thrombocytopenia, renal tubular defect, congestive cardiac failure, arrhythmias, gynecomastia, parathyroid insufficiency etc. Diagnosis is made on the basis of laboratory findings of elevated serum bilirubin, deranged coagulation profile, low serum ceruloplasmin levels, increased 24 h urinary copper levels, increased hepatic copper levels. The brain lesions on MR imaging are frequently seen in the Putamen, Caudate nuclei, Globus Pallidus, Thalamus, Midbrain, Pons, Substantia Nigra, subcortical white matter, Centrum Semiovale, Periaqueductal gray matter, Dentate nucleus, Red nucleus and Vermis. Chelating agents are helpful in excretion of copper from the body. Chelating agents include D-Pencillamine, Trientene, Ammonium tetrathiomolybdate and zinc. Liver transplantation is the treatment of choice in fulminant hepatic failure.
Introduction
Wilson disease is a metabolic disorder with autosomal recessiveinheritance involving a defect in excretion of copper from the bodythat results in accumulation of copper in liver, brain and eyes resultingin their damage, thus called as hepatolenticular degeneration. [1]Gene involved is ATP7B on chromosome 13, which is a coppertransporting gene expressed mainly on hepatocytes and is responsiblefor biliary copper excretion. Mutations in ATP7B can occur anywherealong the entire 21 exons, which makes the identification of genedefects difficult. Identification of carriers and presymptomatic familymembers of affected individuals is achieved by polymerase-chainreaction-based marker analysis [2]. Genetic defect in the form ofeither absence or malfunctioning of ATP7B gene results in decreasedexcretion of copper in bile leading to its accumulation in hepatocytes and when hepatocytes are overloaded there is redistribution toother organs (brain, eyes and kidneys). [3] Copper is taken intohepatocyte via CTR1 (copper transporter 1) on the sinusoidal aspectof hepatocyte. Copper bound to CTR1 binds ATOX1 which is copperchaperone and then it is delivered to Wilson disease protein ATP7B( which is bound to ceruloplasmin) which brings about transportinto trans-golgi and loading into vesicles for transport of copper intobile and excretion from the body. The most common mutation ishistidine to glutamine substitution at amino acid 1069Q (H1069Q).[3] ATP7B is mislocalised to the endoplasmic reticulum consistentwith a failure of the mutant protein to undergo normal transport totrans-golgi apparatus thus leading to copper deposition.
Hepatic changes include microsteatosis, macrosteatosis,increased glycogen in the nucleus and areas of necrosis. In more advanced disease, the changes observed are quite similar to thoseseen in autoimmune hepatitis, such as infiltration by inflammatorycells, piecemeal necrosis and fibrosis. In the brain, most copperis deposited in the basal ganglia, particularly in the putamen andGlobus Pallidus (together called the Lenticular Nucleus). Hemolysisoccurs in Wilson disease as copper has a direct effect on oxidation ofhemoglobin, inhibition of energy-supplying enzymes in the red bloodcell and direct damage to the cell membrane [4].
Clinical features
Hepatic involvement: Patient can present with fulminant hepaticfailure in form of coagulopathy, encephalopathy and Coomb’snegative hemolytic anemia. Cirrhosis associated with splenomegalyand portal hypertension. Hepatocellular carcinoma is rarelyassociated with Wilson disease [4].
CNS involvement: There is widespread involvement of brain with pathologic changes of degeneration and necrosis of the neurons and supporting cells in thalamus, mid brain, globus pallidus, caudate, putamen, cerebrum and cerebellum leading to dysarthria, tremor, lack of motor coordination, drooling, dysarthria, dystonia, and spasticity. Migraine headaches and insomnia have also been reported, although seizures could be more common. Along with behavioral changes, other psychiatric manifestations include depression, anxietyand frank psychosis [4].
Eye changes: Kayser Fleischer (K-F) rings are most apparent at the periphery of the cornea (Figure 1). They are caused by the granular deposition of copper on the inner surface of the cornea in Descemet’s membrane. The upper pole is affected first. The rings have a golden brown appearance. KF ring starts from Schwalbe line and extends till 5mm on corneal surface. It starts from 10 to the 2 o’ clock position. KF ring also starts in the inferior pole. Slit lamp examination is necessary to confirm the presence or absence of K-F rings [4].
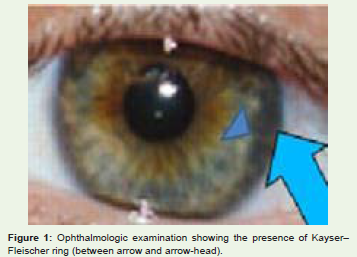
Figure 1: Ophthalmologic examination showing the presence of Kayser–Fleischer ring (between arrow and arrow-head).
Sunflower cataract is the another ocular finding in Wilson disease patients. Sunflower cataract consists of central disc with radiating folds. Sunflower cataract is also seen in intraocular copper bodies (chalcosis), primary biliary cirrhosis and Hereditary hyperferritinemia cataract syndrome [5].
Other eye changes are infrequent or absent blinking, jerkyoscillatory movements of the eye, involuntary upward gaze, nightblindness and pallor of discs and xerophthalmia in isolated cases [6].
Hemolytic changes: Hemolytic anemia due to copper inducedoxidative damage to erythrocytes may be the initial manifestationof Wilson’s disease. Thrombocytopenia has also been seen inWilson disease. [4] Acute hemolytic syndrome can present as initialpresentation of Wilson disease [7].
Renal involvement: Renal tubular dysfunction, with consequenthypercalciuria and hyperphosphaturia, may induce nephrocalcinosis.Hypokalemia with muscle weakness and even respiratory failure hasalso been seen in Wilson’s disease, presumably secondary to renaltubular dysfunction [4].
Skin: changes with hyperpigmentation of the anterior lower legs has been seen in Wilson disease [4].
Gynecological abnormalities (menstrual irregularity, delayed puberty, gynecomastia), cardiovascular dysfunction (congestiveheart failure, cardiac arrhythmia), and other impairments (glucoseintolerance, parathyroid insufficiency) have also been described [4].
Diagnosis
Laboratory findings: Elevated serum bilirubin, reduced serum albumin and coagulation factors [2].
Slit lamp examination: Kayser Fleischer ring may be seen inthe periphery of cornea. The upper pole is affected first. The ringshave a golden brown appearance. Sunflower cataracts are brilliantlymulticoloured and are visible only by slit-lamp examination. Theydo not impair vision. Other less common findings include nightblindness, exotropic strabismus, optic neuritis, and optic disc pallor[7].
Serum Ceruloplasmin levels: Low S. Ceruloplasmin levels < 20mg/dl are consistent with Wilson disease when associated withKF ring. Extremely low S. Ceruloplasmin level < 5 mg/dl is strongevidence of Wilson disease [2].
24 h urinary copper excretion: In Wilson’s disease, the 24 h urinary copper excretion is increased and the concentration taken as suggestive of disease is greater than 100 μg/24 h. Normal level of 24 h urinary copper excretion is < 40 μg/24 h [2].
Hepatic copper levels: Hepatic parenchymal copper content >250g/g dry weight provides critical diagnostic information. In untreatedpatients, normal hepatic copper content (< 40-50 g/g dry weight)excludes a diagnosis of Wilson disease. Further diagnostic testing isindicated for patients with intermediate copper concentrations (70-250 g/g dry weight of liver) especially if there is active liver disease or other symptoms of WD [2].
Neuroimaging
Copper is accumulated in the liver, and once hepatic binding sites are saturated, it is released. Following which, there is development of systemic disease which is accompanied by abnormal deposition of copper in the brain, particularly in the Putamen and GlobusPallidus . [8,9] The neurologic signs and symptoms associated withthis disease are secondary to increase in cerebral copper at levelssufficient to destroy nerve cells. Histopathological changes that areobserved in Wilson’s disease involving the brain includes edema,necrosis, and spongiform degeneration. [10] MR imaging not onlymakes available biochemical information on heavy metal distributionin brain parenchyma but also furnishes an insight into the pathologicand anatomic correlates of clinical signs and symptoms in Wilson’sdisease. [11] Interval changes seen on follow-up MR imaging havean excellent parallel with clinical symptoms, and can be helpful inassessing the clinical response to treatment of children with Wilsondisease [12].
MR imaging findings of the brain in children with Wilson disease can be divided into three diverse groups. [12] In first group, MR imaging findings are normal.
In second group, the basal ganglia are hyperintense on T1-weighted images, which implies hepatic involvement—that is, chronicliver parenchymal disease. In third group, hyperintense lesions areseen on T2-weighted images, which suggests cerebral involvementand show a fine correlation with neurologic symptoms.
The brain lesions are commonly seen in the Putamen, Caudatenuclei, Globus Pallidus, Thalamus, Midbrain, Pons, Substantia Nigra,subcortical white matter, Centrum Semiovale, Periaqueductal graymatter, Dentate nucleus, Red nucleus and Vermis. These lesions areusually bilateral and symmetrical [Figure 2]. The basal ganglia areshow most common involvement followed by the thalami, brainstem,cerebral and cerebellar atrophy.[9] In chronic cases, atrophic changesmay be seen. Hypointense signal intensity on T2- weighted images incorpus striatum and superior colliculus can be appreciated sometimes,due to paramagnetic effects of the copper or iron deposition. [14-15]Sometimes, foci of restricted diffusion can be visualised early in thedisease process due to inflammatory cell swelling due to excessivecopper deposition [16].
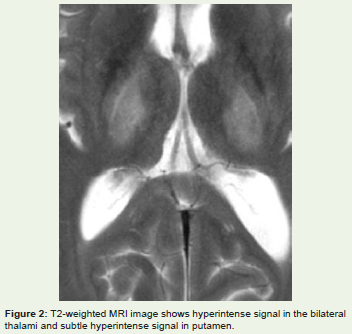
Figure 2: T2-weighted MRI image shows hyperintense signal in the bilateral thalami and subtle hyperintense signal in putamen.
The midbrain “face of the giant panda” sign [14] comprises hyperintense signal in the tegmentum, unchanged signal intensity ofthe lateral portion of the pars reticulata (substantia nigra) and rednucleus (arrowhead), and hypointense signal of the superior colliculus[Figure 3]. Also, a “face of panda cub” [Figure 4] may be visualizedin the dorsal pons. “Eyes of the cub” are created from the relativehypointensity of the central tegmental tracts (CTT) (arrowhead)compared to the hyperintensity of the aqueduct opening into thefourth ventricle (“nose and mouth of the panda”) and boundedinferiorly by the superior medullary velum. The cub’s “cheeks” areformed from the superior cerebellar peduncles [8]. Face of the giantpanda and her cub comprise the “double panda sign” [8] which istypical for this disease.
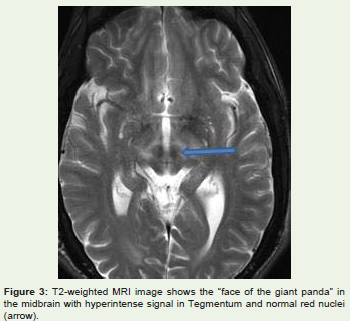
Figure 3: T2-weighted MRI image shows the “face of the giant panda” in the midbrain with hyperintense signal in Tegmentum and normal red nuclei(arrow).
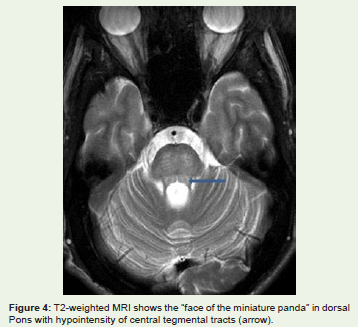
Figure 4: T2-weighted MRI shows the “face of the miniature panda” in dorsal Pons with hypointensity of central tegmental tracts (arrow).
Treatment [17,18]: Anti copper therapy has to be life-long
Chelating agents: Pencillamine is cysteine, doubly substitutedwith methyl groups. A free sulphydryl group acts as the copperchelator.The initial dose of Penicillamine is 1000-1500 mg per dayin two to four divided doses. The treatment is best taken 1 h beforeor 2 h after food. Early side-effects in the first 1-3 weeks include sensitivity reactions with fever, rash, lymphadenopathy, neutropenia,thrombocytopenia, and proteinuria. If these adverse effects arenoticed, then Penicillamine should be stopped and an alternativetreatment used. Later side-effects include nephrotoxicity (a lupuslikesyndrome) and bone marrow suppression (thrombocytopeniaand aplasia). Skin complications have arisen with long-term use ofpenicillamine including progeriatric changes (with long-term dosesgreater than 1000 mg per day), elastosis perforans serpiginosa andaphthous stomatitis.
Triethylene tetramine dihydrochloride (Trien, TETA,trientine) at a dose of 0.5-2.0 g/day for adults and 20 mg/kg/day forchildren.
Ammonium tetrathiomolybdate: It is emerging as the drugof choice because of its rapid control of free copper, preservation ofneurologic function and low toxicity. The initial dose is 120 mg/day(20 mg between meals TID and 20 mg with meals TID). If a nighttimesnack is eaten, another 20 mg is given with the snack. Side effectsinclude anemia, leukopenia, thrombocytopenia and mild elevationsof transaminases.
Zinc impairs the gastrointestinal absorption of copper. Zincacetate is given in adults at a dose of 25–50 mg of elemental zinc threetimes a day and 25 mg three times a day in children >5 yr of age.
Liver transplantation is the only effective treatment for patientswith Wilson disease who have acute liver failure and patients whopresent with decompensated liver disease.
Special conditions
Pregnancy: Chelating agents should be reduced to minimum. Zinc can be continued throughout pregnancy. Breast feeding is not recommended in cases of women taking D-Pencillamine as it may harm the baby.
Conclusion
MRI not only gives biochemical information on heavy metaldistribution in brain parenchyma but also present an insight into thepathologic and anatomic correlates of clinical signs and symptoms inWilson’s disease.
References
- Rebecca G Carey, William F. Balistreri. Metabolic Diseases of the Liver. Kliegman: Nelson Textbook of Pediatrics, 19th ed.; 349: 1391-2.
- Das SK, Ray K. Wilson’s disease: an update. National Clinical Pract Neurol. 2006;2: 482-493.
- Schaefer M, Roelofsen H, Wolters H, Hofmann WJ, Müller M, Folkert Kuipers, et al. Localization of the Wilson‘s disease protein in human liver. Gastroenterology. 1999; 117:1380-1385.
- Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397-408.
- Amalnath DS, Subrahmanyam DKS. Ocular signs in Wilson disease. Ann Acad Neurol 2012;15:200-201.
- Saiduzzafar H, Ansari Z, Kumar M. Wilson's disease with special reference to ocular manifestations (A case report). Indian Journal of Ophthalmology1978;26:37-39.
- Dabrowska E, Jablonskaâ€Kaszewska I, Ozieblowski A, Falkiewicz B. Acute haemolytic syndrome and liver failure as the first manifestations of Wilson's disease. Med Sci Monit. 2001;7(Suppl 1)246-251.
- Jacobs DA, Markowitz CE, Liebeskind DS, Galetta SL. The double panda sign in Wilson’s disease. Neurology. 2003;61:969.
- King AD, Walshe JM, Kendall BE, Chinn RJ, Paley MN, Wilkinson ID, et al. Cranial MR imaging in Wilson’s disease. AJR Am J Roentgenol. 1996;167:1597-1584.
- Harper C, Butterworth R. Nutritional deficiencies and metabolic disorders. In: Greenfield JG, Hume Adams J, Duchen LW, eds. Greenfield’s Neuropathology. 5th ed. London, UK: Edward Arnold; 1992: 838-40.
- Sinha S, Taly AB, Ravishankar S, Prashanth LK, Venugopal KS, Arunodaya GR, et al. Wilson's disease: cranial MRI observations and clinical correlation. Neuroradiology. 2006;48:613-621.
- Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW, et al. MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. AJNR Am J Neuroradiol. 2006;27:1373-1378.
- Yuh WT, Flickinger FW. Unusual MR findings in CNS Wilson disease (letter). AJR Am J Roentgenol. 1988;151:834.
- Hitoshi S, Iwata M, Yoshikawa K. Midbrain pathology of Wilson’s disease: MRI analysis of three cases. J Neurol Neurosurg psychiatry. 1991;54:624-626.
- De Haan J, Grossman RI, Civitello L, Hackney DB, Goldberg HI, Bilaniuk LT, et al. High-field magnetic resonance imaging of Wilson’s disease. J Comput Assist Tomogr. 1987;11:132-135.
- Sener RN. Diffusion MR imaging changes associated with Wilson disease. AJNR Am J Neuroradiol. 2003;24:965-967.
- Roberts EA, Schilsky ML. American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008; 47: 2089-2111.
- Brewer GJ. Wilson’s disease. 18th ed. In: Harrison’s Principles of Internal Medicine. Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J: New York: Mc Graw Hill; 2012: 3188-90.